Neural crest-like cells are what cause glomagenesis to mimic an injury response
Inferring the CNA from scRNA-seq data: using an inferCNV package to identify the clonal regions in our normal brain stem cell dataset
The human GBM stem cell dataset (20 samples) from ref. 53 was downloaded from the Single Cell Data Portal and analysed using the same analysis pipeline and parameters as for our tumorigenesis atlas dataset except with human orthologues. The same quality control cutoffs as for our atlas were used; Seurat and Harmony were used to integrate, correct batch effects, and cluster cells together. For computation of genes that are differentially expressed, the same ComputationalPipeline was used as for the Atlas.
To investigate the clonal distribution of tumour cells in the space, we applied inferCNV to the Visium data. The cells in each Visium spot are assumed to be the same clone, under the assumptions of this analysis. Given the small number of cells contained in each spot, we consider this as a reasonable assumption.
We used the R package inferCNV (v1.7.1), inferCNV of the Trinity CTAT Project (https://github.com/broadinstitute/inferCNV), to estimate the CNAs from scRNA-seq data. The parameters used for 10x genomics data were adopted by us. We picked out three samples from our normal brain atlas as a reference. Running inferCNV resulted in a continuous, gene-level relative CNA profile for each cell. We can see that there are three main peaks, with one large peak and two smaller peaks, which we believe are a gain of copy and loss of copy, respectively. Theoretically, one copy of gain or loss should cause a shift of 0.5 in the relative copy number, meaning that the centre of the peak should be around 1.5 for a gain of copy and 0.5 for a loss of copy. Owing to the high level of noise in scRNA-seq data, the inferred CNAs are far from perfect, presenting a smaller one-copy shift than 0.5. We rounded the two peaks of the inferred CNAs to integers to infer the absolute copy numbers. We first removed genes without clearCNAs. We then identified the two CNA peaks by clustering the CNA values into two clusters using k-means. The one-copy shift in the data can be calculated using the centroids of the two clusters. Then a rescaling factor can be calculated as the fold change between the theoretical one-copy shift (0.5) and the estimated one from the data. After rescaling the CNA profile, we rounded it to integers to calculate the absolute number of copies (round(CNA × 2)).
When estimating the reference cell type signatures, the ‘sample ID’ served as the batch_key to mitigate batch effects. Potential biases are counteracted by the total UMI count and the mitochondrial gene ratio. After these references were established, we performed the deconvolution separately for tumour and normal regions using their respective references. The reference for deconvolution was used for human samples as they were derived from tumour regions.
The one-sided Fisher tests of the proportions of cell types are performed in different time points, and they were computed using the R package rstatix. For each time point, we calculated the number of cells in each cell category to construct the contingency table. The function fisher_test was used to do the tests.
Finer resolution analysis was done on all the cells that were examined, however further clustering was done for only some of the cells.
Raw sequencing data from the 30 scRNA-seq samples (Extended Data Table 1) were aligned to the mm10 mouse reference using Cell Ranger (v3.1.0). Doublets and multiplets were identified by scDblFinder (v1.4.0), and low-quality cells (percentage of mitochondrial >12%; number of genes detected per cell <800; number of unique molecular identifier (UMI) per cell <500) were removed as part of the quality control process. Seurat was used to unify the data from all the samples. We used Seurat’s merge function to combine data from all samples together. The methods used for clustering were the following: Seurat’s LogNormalize method with a default value of 2,000 variables, scaling, rann method for locating nearby cells, and PCA reduction with the first 30 dimensions. Cell type assignment was based on the differentially expressed gene analysis and using the known cell type markers from the literature. We computed the top differentially expressed markers for each of the clusters compared to all other clusters using Seurat’s Wilcoxon rank sum method with a minimum cell fraction of 0.25 and a minimum fold difference of 0.25, and then ranked them by their fold changes and adjusted P values.
Clone identification and directionality of evolution were evaluated from two different perspectives. We used MEDALT and hierarchical clustering with three different distance measures of CNAs, namely the Euclidean distance, the MED and the SD on the tree given by MEDALT. MED was calculated using MEDALT. We loaded the tree given by MEDALT into R as an igraph object, visualized it using GGally::ggnet2() and calculated pairwise SD using igraph::distances(mode = ‘all’).
During cell division and from the parent cell are the ways in which the CNAs in a cell are acquired. We simulated the CNA accumulation on the whole phylogenetic tree we generated. We assume that each cell has a number of newly acquired CNAs. This means that with probability e−λ, a cell will not get any new CNA. In our simulation, λ was set between 0.1 and 0.5. The effect of CNAs is that a gain or loss of copy for adjacent genes can happen together. We assumed that all of the genes that one CNA affected were followed by another random distribution of 100–200 in our simulation. The cells were picked for analysis after each simulation.
The birth–death with immigration model is used. 8), we followed the minimal model of tumour growth proposed in ref. 23. This model is based on a two-component hierarchy, which involves transitions from a stem cell-like compartment to a progenitor population. The simulation is similar to the birth–death model; the difference is that cell division happens in a different way for S cells and P cells. The branching of the P cells is the same as the classic birth–death model with an equal chance of being a death or a birth. The P and S cells will be split asymmetrically if the S cells have high probability. However, with a small probability, it can also divide symmetrically to self-renewal. In our simulation, we assumed that the birth and death rates for P cells and the probability of symmetrical division for S cells do not change during the evolution.
B and d are the birth and death rates in which N0 is the initial number of cells. The interval of time Δt between two adjacent events (the length of the branch in the phylogenetic tree) follows an exponential distribution with mean E(Δt) = 1/(b + d). When branching happens, it can be a birth with a probability of b/(b + d), or a death with a probability d/(b + d). In our simulation, we assumed that the birth and death rates do not change during the evolution.
Cell-cell communication analysis in tumorigenesis using CellChat v1.1.338 and aggregation functions in destiny v3.4.0
We used CellChat v1.1.338 for the analysis of cell–cell communication, which was performed separately for each of the four stages of tumorigenesis. We followed the default parameters of the official process. First, we loaded the normalized counts into CellChat, followed by the preprocessing steps identifyOverExpressedGenes() and identifyOverExpressedInteractions(). We smoothed gene expression by applying a diffusion process on the protein–protein interaction network implemented in projectData() function. We used the computeCommunProb function to get rid of possible bias due to cell population size. This resulted in a network of communication strength between all cell states for each of the ligand–receptor pairs that passed the filtering steps. We used the aggregation functions computeCommunProbPathway() and aggregateNet() to determine the communication strength between cell states at pathway and global levels, respectively. The data slot netP was evaluated on the basis of the role of different cell states as senders or recipients on the basis of the out- degree or in-degree of the communication network.
The slingshot and phateR were used to build the trajectory. We used p hateR to create two-dimensional (2D) embeddeds and 50 harmony embeddeds. Then we built a spanning tree of different cell types in the 2D PHATE space using slingshot. For differential analysis, we first clustered cells into five clusters along the trajectory, and then the cluster-specific maker genes were found using the FinderAllMarkers() function in Seurat. To test the robustness of the results, we also used the diffusion map implemented in the R package destiny v3.4.0 to generate the 2D embeddings of the 50 harmony embeddings, and used PAGA in scanpy to summarize the k-nearest neighbours (knn). All of these methods and others resulted in trajectories with similar topologies between cell states.
Source: Gliomagenesis mimics an injury response orchestrated by neural crest-like cells
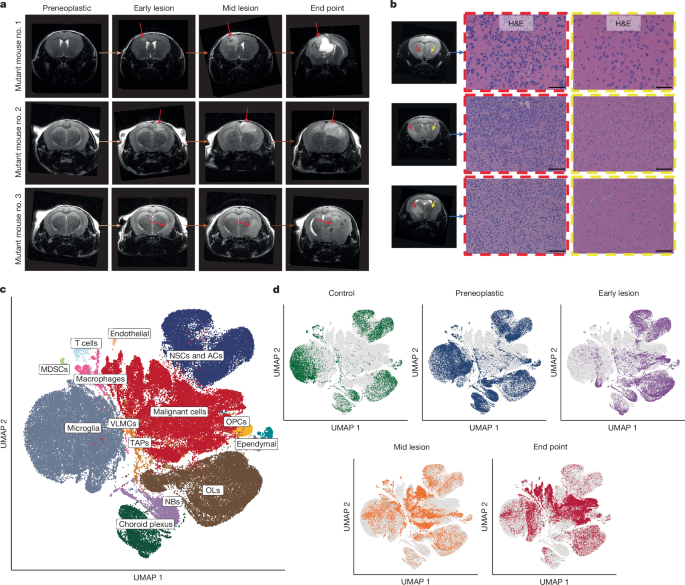
Ethical and legal control of transgenic mouse models for end-point symptoms of focal neuronal abnormality or raised intracranial pressure: A comparative study across four RSTE datasets
The transgenic mice used in this study were obtained from Jackson Laboratories, with the following exceptions: Sox2creER (B6;129S-Sox2tm1(cre/ERT2)Hoch/J) from Konrad Hochedlinger56; Trp53f/f from Chi-chung Hui57; Sox2eGFP (Sox2tm1Lpev) from Freda Miller58. The glioma and control mouse models were kept in a dark and light room with proper temperature and humidity where the mice would have free access to water and chow. Once the mice developed end-point symptoms of raised intracranial pressure or focal neurological abnormality, they were euthanized. The mouse experiments were all performed following the ethical and legal regulations. The experiments and animal use protocols were approved by the Animal Care Committees in the different institutions at the University of Toronto, including the Hospital for Sick Children and University Health Network.
All segmented cells were mapped to the ABC-WMB cell-type taxonomy with the same method used for scRNA-seq data as described above. The high-quality cells of the four RSTE datasets were selected using a combination of thresholds for mapping confidence score and segmentation confidence score. Owing to the variable gene panels and brain regions across the four RSTE datasets, we used a different set of filter criteria for each experiment. For RSTE1, neurons with between more than 50 and fewer than 3,000 transcripts, more than 0.9 average segmentation confidence and three or more unique genes were retained; non-neuronal cells and IMNs between more than 10 and fewer than 1,000 transcripts, more than 0.9 average segmentation confidence and three or more unique genes were retained. Three or more unique genes and a map score greater than 0.4 were retained for RSTE2 cells with between 50 and fewer than 3000 transcripts. For RSTE3, tanycytes with more than 300 transcripts were retained, followed by astrocytes with more than 10 unique genes. For RSTE4, neurons with more than 50 transcripts, more than 0.95 average segmentation confidence and three or more unique genes were retained; non-neuronal cells with more than 20 transcripts, more than 0.95 average segmentation confidence and three or more unique genes were retained. Cell counts for each experiment before and after quality filtering are shown in Extended Data Fig. 5. The analysis was done across the image tiles. A single imaging tile is equivalent to 296 × 296 µm. The ranges of tiles that have been imaged per region are listed.
Identifying the fractions of the malignant cells in an osmotic implanted mouse cannula using FastMnN60 and BBKn61
Mice were prepared for surgery and anaesthetized using isofluorane. The intracranial cannula (Brain infusion kit 3) was implanted at 1.5 mm lateral and 0 mm posterior to bregma, and the saline osmotic pump (Alzet Model 1007D) was implanted subcutaneously following the manufacturer’s instructions. Each osmotic pump was put into a container of normal salt and used to inject the brain at a rate of 0.25 l h1 for 5 days.
In order to ensure downstream analysis is not biased by the integration method, we used FastMnN60 andBBKn61 to redo the batches. These methods all gave similar results (Supplementary Fig. 6).
We first isolated the cells from the malignant cells, and then we removed all of the genes associated with the S and G2/M phases from the mouse orthologues. To see the cycling subtypes, cell cycle genes that were expressed at high levels had to be removed. Reclustering revealed six cycling cell types (cycling NSC-, cycling OPC-, cycling NPC-, cycling MSC-, cycling AC- and cycling NCC-like cells). We calculated the number of cycling and non-cycling cells in each sample to determine the fractions of the malignant states (NSC, OPC, NCPC, NMC, etc.). When calculating the PC-like fractions of cycling and non-cycling cells, the number of cells over the sum of both cycling and non-CYCLING cells was used. We plotted the averages along with their standard errors.
The relativeCNA was stored in the Seurat database and then processed with the log-normalization pipeline with the default parameters. We obtained a new set of UMAP visualization. We observed that most CNA clusters (clones) contain different cell states. We fed the PCA embedded into the miloR v 1.5.064 package and clustered cells into neighbourhoods to elaborate on this. The colour of the cell state in the neighbourhood index cell is indicated by how large or small the neighbourhood is. The number of cells shared between neighbourhoods are depicted in graph edges.
The R package gprofiler2 was used for GO term enrichment. The first and second parts ofv. 0.2.2. The function gost was implemented using parameter ‘ordered = T’ to perform enrichment analysis using a hypergeometric test followed by correction for multiple testing on positive and negative age-DE genes separately. We queried all databases included in gprofiler’s default implementation (GO:molecular function, GO:biological process, GO:cellular component, KEGG, Reactome, TRANSFAC, miRTarBase, Human Protein Atlas, Human Phenotype Ontology). GO terms are only shown in the main figures and not in the Supplementary Table 4. An adjusted P value cutoff of 0.01 was used to determine significant terms. Multiple testing correction was made using gprofiler2’s default program, which accounts for the dependency of multiple tests in the context of enrichment analysis, by taking into account the overlap of functional terms. It’s less strict than the false discovery rate, but is more conservative. The figures have enrichment analysis P values adjusted using this method. GO significance scores are shown in a table. Positive scores were enriched in genes that were increasing with age and negative scores were enriched in genes that were decreasing with age.
where age and sex are all categorical variable each with two levels, and gene detection (gc) and QC score (qc) are log transformed and then z-score normalized, and the tilde (~) means distributed as. We included both genes detection and Quc scores in each model to account for potential effects that different population plans had on library quality. 6e,f). The model with and without the age term had their likelihood ratio computed. Multiple hypothesis testing were done with the corrected P values. The age effect size estimate is found throughout the main body of the text and it is described as thelog2FC of the genes with covariate adjustment.
We observed 2,467 clusters after the first round of clustering. All cells were labeled with the same versions of the ABC-WMB reference taxonomy at this point. The cell annotations of aged cells were assigned on the basis of cluster membership with the annotated adult cells. Specifically, clusters that contained more than 5% of annotated adult cells were assigned that cell category. Median gene detection (GCmed) and median QC score (QCmed) were calculated for each cluster. The data were removed from the clusters that were in non-neuronal and IMN categories. There are clusters that are in the neuronal category with GCmed 3,000. Clusters with more than 80% contribution from a single library were also filtered out to minimize donor bias in the final dataset. The clustering that was less than 5% adult cells were retained and carried over into the next round. Because adult cells that were previously deemed to be low quality were also included in clustering, clusters with the most low-quality cells were also filtered out. In total, 1,197 clusters were removed based on these criteria after the first round of clustering (n = 796,126 cells removed). This resulted in the dataset of 1,203,850 cells, which were carried over into the second round of clustering (Extended Data Fig. 3a).
where a is the number of aged cells in the cluster, b is the number of adult cells in the cluster, c is the number of aged cells in the class minus a and d is the number of adult cells in the class minus b. For tanycytes, b and d were calculated at the subclass level rather than the class level due to inclusion of extra cells from the ABC-WMB atlas that were not included for other cell types.
Cells from Staining Transcript Transcripts – A Baysor Based Approach for Cell Boundary Segmentation in Molecular Cartography
The data shown here has been generated by Resolve Biosciences with their commercially available platform. Four Molecular Cartography experiments were conducted which included each with a different panel of 100 genes and different regions of the brain. For RSTE1, four different regions of the brain can be imaged in both genders and both ages with two replicate brains per condition and two technical replicates per brain. The technical replicates were plotted and analysed as independent replicates in all figures. Four brains were duplicated per condition for the RSP and hippocampus for RSTE2. For RSTE3 and RSTE4, the hypothalamus was imaged in both sexes and both ages, with four replicate brains per condition. Brain dissection and cryosectioning for Molecular Cartography experiments were performed at the Allen Institute for Brain Science in Seattle, WA, samples were stored at −80 °C for 1–3 days, and then shipped overnight to Resolve Biosciences in San Jose, CA, USA where the Molecular Cartography protocol was performed. Spot data was available for 1–2 weeks after receipt of tissue. Data analysis was done at the Allen Institute. There was a way for transcript data to be broken down into different types of cells, as well as a way to determine the quality of the data.
The brains of the fresh-frozen adult and aged were sectioned at 10 m. The OCT block containing a fresh-frozen brain was trimmed in the cryostat until reaching the desired region of interest. Sections were placed onto coverslips provided by Resolve Biosciences. Two replicate sections were collected sequentially: one as the primary sample and the other as a backup.
Cells were segmented using a combination of open-source software Cellpose92 (v.2.1.0) and Baysor93 (v.0.6.2). Cellpose uses a generalist algorithm for segmenting cells from images of cellular stains as input. Baysor uses a transcript-driven algorithm to draw cell boundaries based on transcript data alone while also having the option of integrating previous knowledge from stained images into the process. First, images of DAPI stains from each of the tissue samples were used as input for Cellpose using the following parameters: –pretrained_model = nuclei, –diameter = 0. The output of Cellpose was saved and used to create a prior for the Baysorsegmentation algorithm. Baysor was run with the following input parameters: -m 30, -s 50.
We used a strict process to remove low-quality cells. The first quality cut-off to be used for removing cells was based on genes detection, QC score and doublet score. A set of genes with lower expression levels in poor-quality cells are the ones that are summing up the log- transformed expression of a set of genes. The genes are strongly expressed in nearly all cells and are anticorrelated with the nucleus enriched transcript Malat1. We use this QC score to quantify the integrity of cytoplasmic messenger RNA (mRNA) content. Doublets were identified using a modified version of the DoubletFinder algorithm89. We included cells with gene detection greater than 1,000, a QC score greater than 50, and a doublet score less than 0.3, for this preliminary round of filters. More than a thousand,000 cells remained in the dataset using the thresholds. Mixing of cell types by library and other metadata categories is visualized in Extended Data Fig. 6f.
The Reagent Kit v.3 is used for 10xv3 processing. We followed the manufacturer’s instructions for cell capture, barcoding, reverse transcription, complementary DNA amplification and library construction. We targeted a sequencing depth of 120,000 reads per cell; the actual average achieved was 77,743 ± 36,025 (mean ± s.d.) reads per cell across 287 libraries (Supplementary Table 1).
Cells were collected using a 130 m nozzle and used to enrich for live cells. Cells were prepared by passing the suspension through a 70 m filter and adding HOechst to the final concentration. The strategy was used with the tdTomato- positive label and most cells were collected. 30,000 cells were sorted into a tube containing 500 l of quenching buffer. We found that sorting more cells into one tube diluted the ACSF in the collection buffer, causing cell death. We also observed that cell viability decreased for longer periods. Each aliquot of sorted 30,000 cells was gently layered on top of 200 µl of high BSA buffer and immediately centrifuged at 230g for 10 min in a centrifuge with a swinging bucket rotor (the high BSA buffer at the bottom of the tube slows down the cells as they reach the bottom, minimizing cell death). We removed the supernatant and left behind a 35 l buffer so we could resuspended the cells. Immediate centrifugation and resuspension allowed the cells to be temporarily stored in a high BSA buffer with minimal ACSF dilution. The resuspended cells were stored at 4 °C until all samples were collected, usually within 30 min. Samples from the same ROI were pooled, cell concentration quantified and immediately loaded onto the 10x Genomics Chromium controller.
Tissue pieces were taken out of their body with 30 U grams of papain. PAP2) in ACSF for 30 min at 30 °C. We decided to change the oven temperature to 35 C due to the short amount of time in the oven, but keep the solution temperature at 30 C. Enzymatic digestion was quenched by exchanging the papain solution three times with quenching buffer (ACSF with 1% fetal bovine serum and 0.2% bovine serum albumin (BSA)). The samples were put on ice for 5 minutes before trituration. The tissue pieces in the quenching buffer were triturated through a fire-polished pipette with a 600 µm diameter opening roughly 20 times. The tissue pieces were allowed to settle and the supernatant, which now contained suspended single cells, was transferred to a new tube. Fresh quenching buffer was added to the settled tissue pieces, and trituration and supernatant transfer were repeated using 300 and 150 µm fire-polished pipettes. The single-cell suspension was passed through a 70 µm filter into a 15 ml conical tube with 500 µl of high BSA buffer (ACSF with 1% fetal bovine serum and 1% BSA) at the bottom to help cushion the cells during centrifugation at 100g in a swinging bucket centrifuge for 10 min. The supernatant was discarded, and the cell pellet was resuspended in the quenching buffer. We collected more than one million cells. The concentration of the resuspended cells was quantified, and cells were immediately loaded onto the 10X Genomics Chromium controller.
The single cells were isolated from each other. The brain was dissected, submerged in artificial cerebrospinal fluid (ACSF), embedded in 2% agarose and sliced into 350-μm coronal sections on a compresstome (Precisionary Instruments). The images were taken during the slicing process. ROI were then microdissected from the slices and dissociated into single cells. Fluorescent images of each slice before and after ROI dissection were taken at the dissection microscope. These images were used to document the precise location of the ROI using annotated coronal plates of CCFv3 as reference.
The Allen Institute for Brain Science used the Institutional Animal Care and Use Committee protocols to carry out procedures. Food and water was provided to the rodents and they were kept in their cages with no more than five animals of the same sex per cage. The temperature and humidity in the mausoleum was maintained between 40 and 45%. The C57BL/6J background has mice on it. We excluded any mice with dermatitis, anophthalmia, microphthalmia, seizures or abdominal masses.
The 64 young adult mice we used to collect cells for 10xv3 scRNA-seq were aged from 18 to 33 years old. All young adult mice were also included in the ABC-WMB atlas3. Aged animals were euthanized at P540–553 (roughly 18 months) and young adult animals were euthanized at P53–69 (roughly 2 months). No statistical methods were used to predetermine sample size. All donor animals used in this study are listed in Supplementary Table 1. The Zeitgeber time of the light/dark cycle was similar (within a 3 h window) for all the tissue collections. We did not keep a record of the female mice’s oestrous cycle.
We isolated a total of 287 libraries from 108 animals: each animal contributed 1–6 libraries. All libraries are listed in Supplementary Table 1. Transgenic driver lines were used for fluorescence-positive cell isolation by FACS to enrich for neurons. Roughly half the libraries (n = 145) were sorted for neurons from the pan-neuronal Snap25-IRES2-Cre line (JAX strain no. 023525) crossed to the Ai14-tdTomato reporter. (Supplementary Table 1). We used snap25- IRES2/Cre/wt, ani14/wt mice, and a few wild type C57BL/6J mice in unbiased sampling. libraries stained and sorted for Hoechst+, Calcein+/Hoechst+ or unstained libraries that were not sorted are excluded from biased sampling methods. No FACS cells make up roughly 25% of the final high-quality dataset. For at least ten generations, the transmogrified C57BL/6J line was backcrossed and can be considered congenic. The Ai14 line was backcrossed into C57BL/6J for at least five generations before being considered congenic. Ging strategies used for FACS are shown in an example.
We used the CCFv3 (RRID: SCR_002978) ontology22 (http://atlas.brain-map.org/) to define brain regions for profiling and boundaries for dissection. We covered all the brain regions by sampling at the top-ontology level. These choices were guided by the fact that microdissections of small regions are difficult. Joint dissection of the adjoining regions is necessary to get enough cells for profiling.
The first-ever cluster-centric analysis of brain stem cell datasets using the inferCNV package was used to identify clonal regions. Raw sequencing data from the 30 scRNA-seq samples and low-quality cells were aligned to mm10 mouse reference using Cell Ranger. Doublet and multiplets were identified by scDblFinder, and low-quality cells were removed as part of the quality control process.