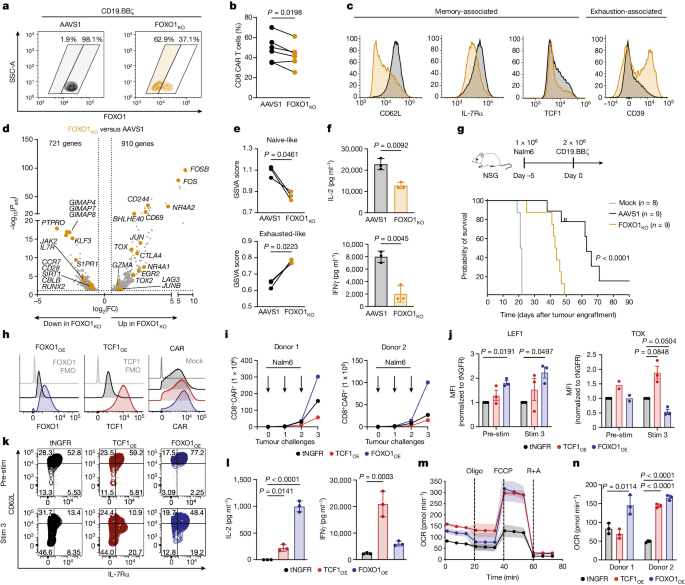
CAR T cell stemness and metabolism are improved by FOXO1
Test of Mycoplasma Using Cryopreserved CD3+, Anti-GD2 and Nalm6 B-ALL T Cells from a Donor Blood Leukopak
For experiments completed at Stanford, buffy coats from anonymous, consenting healthy donors were obtained from the Stanford University Blood Center under an University Institutional Review Board-exempt protocol or obtained from a human peripheral blood leukopak (STEMCELL Technologies). The RosetteSep human T cell enrichment kit and Lymphoprep medium were used for the isolated CD3+ cells. For experiments completed at the Children’s Hospital of Philadelphia (CHOP), purified CD3+ healthy donor T cells were obtained from the University of Pennsylvania Human Immunology Core. All purified T cells were cryopreserved in CryoStor CS10 medium (STEMCELL Technologies).
Cell lines were obtained from ATCC and stably transduced to express markers as follows: 143B osteosarcoma cells express GFP and firefly luciferase with or without CD19, Nalm6 B-ALL cells express GFP and firefly luciferase with or without GD2. The clones were chosen because of their high expression. The cells were fed with 10% fetal bovine serum, 10 mM HEPES, 1 penicillin and other ingredients, cultured in the modified Eagle’s medium. These cell lines, which were engineered prior to this study, had been previously authenticated via STR and have now been used in this study. The National Cancer Institute was the source of the HEK293 cells. The Lonza MycoAlert Mycoplasma Detection kit was used to test cells for mycoplasma.
The CAR constructs used in this study include CD19.28ζ, CD19.BBζ, anti-GD2 HA.28ζ and Her2.BBζ. Gene blocks were created as a result of Codon’s contributions to FOXO1 and P2A ribosomal skip sequence. The tNGFR-only construct does not contain a P2A ribosomal skip sequence. The FOXO1DBD construct was generated by two-step mutagenic NEBuilder HiFi DNA Assembly (New England BioLabs). All plasmids were amplified by transformation into a coliform, and the sequence of them was also checked.
T cells were transduced with retrovirus on days 2 and 3 after activation for all experiments. In brief, 12- or 24-well, non-tissue-culture-treated plates were coated with 1 ml or 500 μl, respectively, of 25 μg ml−1 Retronectin (Takara) in PBS and placed at 4 °C overnight. Plates were washed with PBS then blocked with bovine albumin the next day. PBS for 10 minutes. Retroviral supernatants were added and plates were centrifuged at 32 °C for 2 h at 2,500g. Viral supernatants were subsequently removed and T cells were added to each virus-coated well at a density of 1 × 106 T cells per well for 12-well plates and 0.5 × 106 T cells per well for 24-well plates.
A total of 5 104 CART cells were cultured with 5 104 tumour cells without IL-2 in a 96-well plate. Twenty-four hours after co-culture, culture supernatants were collected, diluted 20- to 100-fold and analysed for IL-2 and IFNγ using ELISA MAX kits (BioLegend) and Nunc Maxisorp 96-well ELISA plates (Thermo Fisher Scientific). Absorbance readings were collected on a Tecan Spark plate reader or a BioTek Synergy H1 running Gen5 v.2.00.18. For FOXO1i assays, the co-culture medium included concentrations of AS1842856 that were used during T cell expansion.
CD8+tNGFR+ CAR T cells were isolated using the EasySep Human CD8+ T Cell Isolation Kit. A total of 150,000 CD8+ T cells were slow-frozen in BamBanker (Bulldog Bio) cell preservation medium. Ten thousand CAR T cells were washed in ice-cold PBS and then subjected to nuclei isolation with the following lysis buffer. NaCl, 3 mM MgCl2, 0.1% Tween-20, 0.1% NP40, 0.01% Digitonin and 1% BSA. The cells were washed and the 50 llysis buffer added to each sample. Nuclear pellets were centrifuged and resuspended in the transposase reaction containing 10.5 μl H2O, 12.5 μl 2× TD buffer and 2 μl Tn5 transposase in a total of 25 μl. The reaction was incubated for 30 min at 37 °C. The response was stopped by the addition of 75 lTE buffer, followed by column purification per the manufacturer’s recommendation. DNA was eluted from the columns in 22 μl H2O. PCR reactions were set up as follows: 21 μl DNA, 25 μl Phusion master mix (NEB) and 2 μl of each barcoded PCR primer (ApexBio, K1058). Each sample had a number of cycles run for it. Reactions were cleaned up with AMPure XP beads according to the recommendations of the manufacturer. Libraries were quantified with a Qubit fluorometer and fragment analysis was performed with Bioanalyzer. The libraries were mapped on the NovaSeq 6000.
To interrogate the role of endogenous FOXO1 in CAR T cell function, CRISPR–Cas9 was used to delete a sequence directly upstream of the FOXO1 DNA-binding domain. CAR T Cells were removed from the beads by magnetic separation on the fourth day after activation. Twenty-microlitre reactions were prepared by injection of the one million CAR T cells in the P3 buffer immediately before releasing them on the P3 primary cell 4D kit. Ribonucleoproteins were prepared by complexing 0.15 ng of sgRNA targeting FOXO1 or AAVS1 (Synthego) with 5 µg Alt-R S.p. There’s a question about the number. Nuclease (IDT, 1081058) before adding the cell suspension to each reaction. The previously verified sgRNA sequence was used for the AAVS1 edits. For FOXO1, two separate sgRNAs were used in tandem, at equal concentrations (5′-UUGCGCGGCUGCCCCGCGAG-3′ and 5′-GAGCUUGCUGGAGGAGAGCG-3′). For TCF7 gene editing, we used a previously validated sgRNA56 (5′-UCAGGGAGUAGAAGCCAGAG-3′) for bulk RNA-seq experiments performed at CHOP. There is a separate sgRNA that was designed and used for experiments. The Lonza 4D Nucleofector used to pulsed the reaction. Cells were recovered immediately in 260 µl of warm complete AIM-V medium supplemented with 500 U ml−1 IL-2 in round-bottom 96-well plates and expanded into 1 ml fresh medium within 24 h. For functional and phenotypic characterization, cells were maintained at a rate of 1.0106 cells per million of volume in well plates. On days 14–16, knockout efficiency was determined by intracellular transcription factor staining (Cell Signaling, 58223) followed by flow cytometry.
T cells were co-cultivated with tumors at a ratio of 2:1 for 24 hours. Fc-receptors were blocked with the human Fc Block (BD BioSciences) for 10 min at 4 C before being stained with the 50 l fluorochrome-conjugated antibodies for 30 min in the dark. Samples were labelled with anchor lipid-modified oligo (LMO) (5′-TGGAATTCTCGGGTGCCAAGGgtaacgatccagctgtcact-[lipid]-3), co-anchor LMO (5′-[lipid]-AGTGACAGCTGGATCGTTAC-3′) and sample specific barcodes for 5 min in the dark at 4 °C. CAR+ T cells were sorted by FACS and samples were pooled at equal ratios followed by staining with 100 μl TotalSeq-C anti-human CD4 and CD8 (BioLegend) antibody cocktail for 30 min in the dark at 4 °C. scRNA-seq data was created using Cell Ranger and hg38 genome. The raw feature barcode matrices were generated using cellranger multi. Downstream analysis was performed in R (version 4.2.0). Doublets were detected and removed using the DoubletFinder function from the DropletUtils (version 1.16.0) package, while empty droplets were detected and removed using the emptyDrops function. Cells with less than 200 features and more than 5% of their reads were excluded from using Seurat. Normalize data, FindVariable features, ScaleData, RunPCA, Run UMAP, FindNeighbors and FindClusters were used to remove clusters with low-quality metrics. The LDOs were demultiplexed. DEGs were calculated using the functions The Find AllMarkers formula uses a log2transformed fold change threshold and a P value less than 0.05, and includes the number of counts as a variable. The run_de function was used to detect pseudobulk DEGs. Gene-set enrichment was performed using the fgsea package with all expressed genes as the background gene list, which was ranked by average log-transformed fold change detected with FindMarkers using a log2-transformed fold change threshold of 0 and min.pct parameter set to 0. To perform diffexp analyses and GSEA between individual groups within each cluster, the to_psuedobulk function from Libra was used to pull out pseudobulk count matrix of each replicate pool and clusters. Gsea and EdgeR were used to perform the differential expression and gsea analyses. The single-cell signature explorer program was utilized for visualization of gene signatures across UMAP plots58.
tNGFR+ cells were isolated and CAR T cells were activated and transduced. Cells were cultured in AIM-V with IL-2 for 14 days before they were tested for stem cells, which is called pre-stim. A final concentration of 5 105 total cells perml was achieved for the co-cultures on day 14. On day 3 of the repeat stimulation co-culture, CAR T cells were again assayed by cytokine secretion, IncuCyte killing assay and flow cytometry as described above. In order to repeat the process for four more co-cultures, the cytokine and IncuCyte assays were set up for four serial stimulations on the day after the first stimulation, 14, 17 and 20. T cells were analysed by flow cytometry on the 7th day of co-culture and on the 14th, the 17th and the 18th day, respectively.
The Sea Horse Bioscience Analyzer was used for the analyses. 0.1 106 cells were resuspended in the assay medium and plated on a Cell-tak microplate with 11M glucose, 2mM glutamine, and 1 mM sodium pyruvate. Mitochondrial activity and glycolytic parameters were measured by the oxygen consumption rate (OCR) (pmol min−1) and extracellular acidification rate (ECAR) (mpH min−1), respectively, with the use of real-time injections of oligomycin (1.5 M), carbonyl cyanide ptrifluoromethoxyphenylhydrazone (FCCP; 0.5 M) and rotenone and antimycin (both at 0.5 M). The manufacturer’s instructions were used to calculate respiratory parameters. Reagent sources are listed in Supplementary Table 2.
Chromatin-bound and soluble proteins were separated as previously described23. In brief, cytoskeletal (CSK) buffer was prepared using 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 10 mM PIPES (pH 6.8), 0.1% IGEPAL CA-630, 4 µg ml−1 aprotinin, 10 µg ml−1 leupeptin, 4 µg ml−1 pepstatin and 2 mM PMSF. After 20 minutes on ice, the cells were lysed with CSK buffer. The samples were separated from the rest by centrifugation at a rate of 15,870g for 10 minutes. The protein concentration of the soluble fraction was determined by DC protein assay (Bio-Rad, 5000116). The remaining pellet containing the chromatin-bound fraction was washed twice with CSK buffer, centrifuging at 1,500g for 5 min. Chromatin-bound proteins were resuspended in CSK buffer and 1× Pierce Reducing Sample Buffer (Thermo Fisher Scientific, 39000) and boiled for 5 min for solubilization. The soluble fraction was supplemented with Pierce Reducing Sample Buffer to achieve 1× and boiled for 5 min. For immunoblotting, equal amounts of soluble and chromatin-bound fraction for each sample were analysed by SDS–polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes (Bio-Rad, 1704158). Membranes were blocked for 30 min in 5% milk in TBST (1× Tris-buffered saline containing 0.1% Tween-20). After washing with TBST, membranes were incubated with anti-FOXO1 antibody (1:1,000; Cell Signaling, 2880, clone C29H4) overnight at 4 °C. Next, membranes were washed with TBST and incubated with anti-mouse (1:10,000, Cell Signaling, 7074) or anti-rabbit (1:10,000, Cell Signaling, 7076) IgG conjugated to horseradish peroxidase for 1 h at room temperature. Membranes were visualized using Clarity Western ECL Substrate (Bio-Rad, 1705060) and the ChemiDoc Imaging System and Image Lab Touch Software v.3.0 (Bio-Rad). After visualization, membranes were stripped using a mild stripping buffer (1.5% glycine, 0.1% SDS, 1% Tween-20, pH 2.2). The previous steps were repeated for detection of soluble (1:5,000 GAPDH; Cell Signaling, 97166, clone D4C6R) and chromatin-bound (1:1,000 Lamin A; Cell Signaling, 86846, clone 133A2) fraction loading controls. Densitometry analyses were performed using Fiji v.2.14.0/1.5 f.
Ethical characterization of HER2 transgenic mice bred in the Peter MacCallum Cancer Centre using a sterile 70m cell strainer
C57BL/6 wild-type mice and C57BL/6 human-HER2 transgenic mice47 were bred in the Peter MacCallum Cancer Centre animal facility. NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice were either bred at the Peter MacCallum Cancer Centre or obtained from Australian BioResources. Mice used in experiments were between 6 to 16 weeks of age and were housed in PC2 specific pathogen-free conditions and a minimum of 3 mice per group were used in each experiment. Mice were randomized prior to treatment according to tumour size to ensure all groups had equivalent tumour burden prior to therapy. It is not possible to blinded experiments as the same investigators analysed and performed the experiments. The animal experimentation ethics committee approved the experiments. The projects stated ethical endpoints, such as maximum tumours size, and all experiments complied with them.
Retro-orbital blood collection was used to collect peripheral blood from live mice. The fifty microlitres of blood was analysed after being labelled with surface antibodies, lysed and quantified using Count Bright Counting Beads. For phenotypic analysis of spleen and tumours, mice were euthanized and tissues were mechanically dissociated and washed twice in PBS. Spleens were placed in a 6-cm Petri dish and filtered through a sterile 70-µm cell strainer. Tumours were mechanically and chemically dissociated from the rest of the body at a temperature of 37 C and then stirred for 30 minutes. After being mashed through the sterile 70m cell strainer, cells were washed with PBS. Cells from both the spleens and the tumours were treated with lysis buffer for 3 minutes on ice. The cells were washed multiple times and isolated using the EasySep Release Human CD45 positive selection kit. Cells were stained for markers of interest and analysed on an instrument called the Cytek Aurora.
A total of 0.5 × 106–1 × 106 T cells were pelleted by centrifugation and flash-frozen. The pellet was thawed on ice and processed using either an RNEasy Plus Mini Kit or an AllPrep DNA/RNA Micro Kit according to instructions from the manufacturer. The totalRNA was quantified using either a Qubit Fluorometer or the DeNovix DS-11 fx lab instrument, and there were around 50 million read pairs per sample.
We processed the sequencing data using the nf-core RNA-seq pipeline (https://nf-co.re/rnaseq). In brief, we performed quality control of the fastq files using FastQC and trimmed the filtered reads with Trim Galore software. The trimmed fastq files resulting from the experiment were aligned to the hg38 human genome using STAR. The salmon was then used to create a matrix for analysis. PCA was done on read counts that were processed, and plots were created from the top 1,000 variable genes. In order to correct for donor effects the removeBatchEffect function was used. Differential analysis of gene expression was performed using the DESeq2 v.3.16 package, with an absolute log2-transformed fold change ≥0.5 and false discovery rate (FDR) < 0.05. differential genes were aggregated and expressions were standardized to create a heat map. The k-means clustering algorithm with Pearson correlation as the distance metric was used to cluster the genes. Pathway analysis of the differential genes and grouped genes in the heat map was performed using QIAGEN Ingenuity Pathway Analysis 2022 Winter Release and clusterProfiler v.4.6.2. Cell-type enrichment was performed using the GSVA v.1.46.0 R package using signatures from previous studies.
To generate single-cell RNA-seq libraries of tumour-infiltrating CAR T cells, Her2+ tumours were collected from five mice per condition, and human CD45+ cells were isolated by NGFR selection as described above (see ‘Cell selection’). The CAR T cells were further purified with the aid of the Aurora Cell Sorter. There were over 20,000 CAR TILs pooled across five mice. Cells were barcoded and sequencing libraries were generated using the 10X Chromium Next GEM Single Cell 3’ v.3.1 kit (10X Genomics) according to the manufacturer’s instructions. Libraries were sequenced at the CHOP High Throughput Sequencing Core on an Illumina NovaSeq 6000 with an average read depth of 50,000 reads per cell.
The libraries were processed with the default options. Fastq files were pre-aligned to the mitochondria genome so that they couldn’t read theMitochondrial reads. The multi mapping reads that aligned to repetitive regions of the genome were removed from the dataset. Bowtie2 was then used to align the reads to the hg38 genome. SAMtools was used to identify uniquely aligned reads, and Picard was used to remove duplicate reads. The resulting deduplicated and aligned BAM file was used for downstream analysis. Peaks in individual samples were identified using MACS2 and compiled into a non-overlapping 500-bp consensus peak set. In brief, the peaks were resized to 500 bp width and ranked by significance. The peaks that overlapped with the same region were selected by ranks and the most significant peak was retained. The peak-sample count matrix was generated using ChrAccR with the default parameters of the run_atac function. Signal tracks for individual samples were generated within the pepatac pipeline. The tracks were merged into a single group that could see the data across all samples.
The DESeq2 v.3.16 package was used to identify peaks across different conditions, with a threshold of an absolute log2-transformed fold change greater than 5 and P value less than 0.05. Adjusted P values were not used owing to donor variability. To generate PCA plots, we first extracted a variance-stabilized count matrix using the vst function in DESeq2. Next, we corrected for batch effects by donor using the removeBatchEffect function in the limma library. Finally, we generated PCA plots using the corrected matrix with the plotPCA function using the top 2,000 most variable peaks. We aggregated differential peaks across conditions, standardized the peak signals, and performed k-means clustering to generate a heat map. Homer and findMotifsGenome.pl were used to find the enrichment of differential peaks and grouped peaks. The enrichment of cell-type specific regulatory elements was performed using the gchromVAR package. In brief, this method weights chromatin features by log2-transformed fold changes of cell-type-specific regulatory elements from a previous report9 and computes the enrichment for each cell type versus an empirical background matched for GC content and feature intensity.
Downregulated differential genes were intersecting in FOXO1KO cells, and upregulated differential genes were intersecting in the FOXO1 regulon gene set. Regulon enrichment scores were calculated using ssGSEA in the GSVA R package on a previous RNA expression dataset2.
For regulon analyses of single-cell ATAC-seq data, the processed Signac data objects of CAR T products profiled by single-cell ATAC-seq were obtained from a previous study5. To account for sample-to-sample variability, the mean fragments in peaks per cell were downsampled for consistency between donors. The donors were excluded from being included based on low data quality after an examination of quality control statistics. We computed a per-cell epigenetic signature for T cells using the chromVAR Workflow as described in the previous paragraphs. We ran an ordinary least squares regression to find differences in responses and non-responses with this signature. Statistical significance was based on the Wald test statistic of the coefficient for the responder term in the two regressions for each factor.
Unless otherwise stated, statistical analyses for significant differences between groups were conducted using one- or two-way analysis of variance (ANOVA) with Bonferroni, Tukey’s or Dunnett’s multiple comparisons test, or with a Student’s or Welch’s t-test using GraphPad Prism v.9.4.1. In experiments in which same-donor samples were compared across two conditions, we performed a paired Student’s t-test. Survival curves were compared using the log-rank Mantel–Cox test. Statistical methods were not used to predetermine sample sizes.
This research and all study protocols have been approved and comply with the Peter MacCallum Animal Experimental Ethics Committee (AEC) ethical regulations regarding the use of animals. The Peter MacCallum Cancer Centre Human Research Ethics committee approved studies using human peripheral blood mononuclear cells from healthy donors. Informed consent was obtained from the Australian Red Cross.
The isotype control (2A3 clone, IgG2a) and Mouse IFN antibody were purchased from BioXcell. The cytokine IL-2 was obtained from the National Institutes of Health and purchased from Peprotech. IL-7 and IL-15 were purchased from Peprotech. CAR T cells are stimulated with a custom made anti-idiotype antibody where indicated.
There are human nerve growth factor regulatory genes that are clones of mouse TCF7, FOXO1 and ID3 found in the mouse stem cell virus. The anti-HER2 CAR retroviruses were created by a viral packaging GP+E-86 cell line. The anti-HER2 CAR construct was comprised of an extracellular scFv specific for human HER2, an extracellular CD8 hinge region, a CD28 transmembrane domain and an intracellular CD3ζ domain. The generated anti-HER2 CAR cell lines were sorted by the expression of NGFR and mCherry in the cells. CAR T cells were maintained in the RPM supplementedI medium with IL-7 and -mer captoethanol, as well as with supernatants from these cells.
For experiments utilizing human anti-Lewis Y CAR T cells, NSG mice were injected with 5 × 106 OVCAR-3 tumour cells. The mice were treated with 1 Gy total body irradiation and 2–5 106 flag+ after the tumours were established. CAR T cells. Mice were treated with IL-2 as per experiments in the C57BL/6 human-HER2 transgenic model.
GraphPad Prism Analysis of Student’s t-test, One-way and Two-way ANOVA for Multi-dataset comparisons
The analyses were done using GraphPad Prism. Analyses performed include paired or unpaired Student’s t-test to compare two datasets, one-way ANOVA to analyse multiple datasets across a single timepoint and two-way ANOVA when analysing multiple sets of data across time.
Researchers from the University of Pennsylvania have isolated human CD4 and CD8 T cells from healthy donors and isolated human CD45+ cells from NSG mice. A total of 1.5 lakh CD5+ cells were slow-frozen in BamBanker medium. Ten thousand CAR T cells were washed in ice-cold PBS and then subjected to nuclei isolation with the following lysis buffer.