Single cell histone modifications are used for embryo tracing
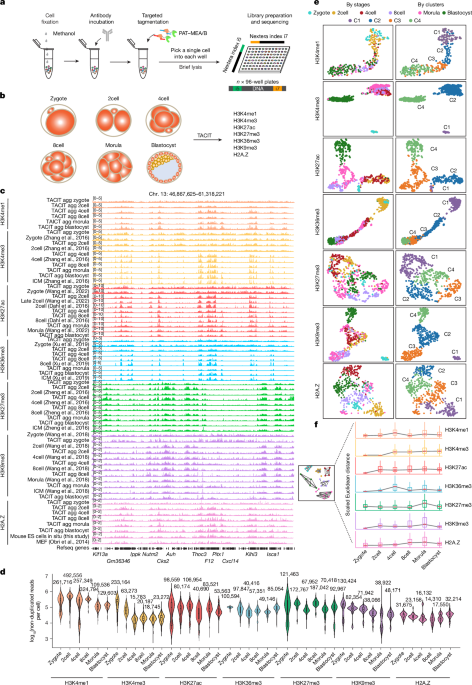
Sequencing and isolation of embryo-barcoded TACIT with 2 cell embryos using pre-warmed Tyrode’s acidic solution for 30 s
For embryo-barcoded TACIT with cells of early and late 2cell stages, the zona pellucida of embryos was gently removed by treating with pre-warmed Tyrode’s acidic solution (Sigma) for 30 s. The embryos were transferred to M2 medium and directly fixed with methanol as described above. The embryos were stored at −20 °C or immediately used. Whole embryos were directly applied to the regular TACIT pipeline as described above. After the embryo was tagged, the two cells from it were separated using needles and deposited into different wells of lysis buffer. Each well was covered with 5 μl mineral oil (Sigma) and incubated at 55 °C for 15 min to release DNA fragments. Next, 0.5 μl of 10 mM Adding a single l of 1% Triton X 100 was added to quench the reaction. Two cells from the same 2 cell embryo were barcoded with different Nextera i5 and i7 indexes after the amplification of their genomic material, as described in the libraries. Finally, the embryo-barcoded TACIT library was sequenced as described for conventional TACIT.
The sgRNAs targeting the promoter were inserted into the CROP-opti vector from the synthesized sgRNAs. Three libraries of sgRNAs included candidate TFs (CEBPG, LBX1, ETS2, MEF2D, ESR2, ESR1 and ALX1), positive control TFs (ZSCAN4 and DUX) and a non-targeting control as previously described53 at equal molar ratios. The lentivirus supernatant was collected 18 h after transfection. The mouse ES cells were infected (8 μg ml–1 polybrene) with various titres of lentivirus to achieve different multiplicity of infection values. New culture medium with 1 puromycin was added for 48h after the first 24 h. Cells were collected for analysis. The cells were preserved at 80 C after being fixed with 1% formaldehyde for 10 minutes. As previously described54, single-cellRNA-seq for mouse ES cells for capturing both sgRNAs was done as part of the same SPLiT-seq pipeline.
For the enrichment analysis in Fig. The expected probability was calculated by taking the length of the total bins and dividing that by the length of the transposable elements in the mouse genome. In 2,583 totipotency- related bin we found 75 transposable elements that were highly enriched. We defined a promoter, enhancer,gene bodies, Heterocronin, or quiescent/low regions as the percentage of transposable elements that were enriched in the cell. To quantify the expression levels of these transposable elements, we mapped raw scRNA-seq reads to themm10 genome using Hisat2 and Samtools. We calculated the numbers of TPM based on the locations of annotated repeats (RepeatMasker) downloaded from the UCSC Genome browser.
Source: Genome-coverage single-cell histone modifications for embryo lineage tracing
Importance of Histone Modifications for the Identification of Interactions in Two-Cell TACIT Profiles using the Random Forest Training Model 62
The late 2cell stage allvalid pairs matrix was downloaded from the GEO database. We used the analyze HiC function of Homer and the python to identify interactions.
We integrated H3K4me1, H3K4me3, H3K36me3 and H3K27ac TACIT profiles with gene expression. The matrix for each histone modification wasgenerated using cisTopic58. The GeneActivity function of Seurat (v.4) was used to create a gene-activity score matrix based on the cell–peak or cell–bin matrix. FindTransferAnchors is used in the identification of the anchors between the two modalities. In particular, many titrations were performed to obtain the highest prediction score, including using the cell–peak or cell–bin matrix, or the bin size of the cell–bin matrix. The cells that had a prediction score lower than 0.2 were removed. Notably, for integrating cells in the 2cell stage, histone-modification signals in non-canonical broad binding regions were excluded before Seurat integration (5 kb for H3K4me3 and H3K27ac, 20 kb for H3K36me3 and H3K4me1).
The two strategies that we used were to rule out differences in stage and to identify the bins between 2cell 1 versus 2cell 2. For each strategy, a cell–bin probability matrix was first generated for each chromatin state, and genomic regions for which posterior probability exhibited a correlation of more than 80% or less than −50% with the expression of totipotency marker genes was selected. Next, all highly correlated candidate bins were aggregated in order to generate a state matrix. The state matrix of synthetic cells at the 2cell and 8cell stages was used as input for constructing the random forest training model62, with labels as ‘toti-high’ and ‘toti-no’ groups, respectively.
The alignment files were converted to a matrix of rows and cells withgenomic intervals using cisTopic. For different histone modifications, different sizes of genomic intervals were used as follows: 5 kb bins for H3K27ac; 10 kb bins for H3K4me3, H3K36me3 and H3K27me3; and 15 kb bins for H3K4me1 and H3K9me3. Seurat was used to cluster the cells based on histone modifications. The normalized cell–bins matrix was first normalized with the term Frequency–inverse document Frequency, followed by Density Reduction with singular value decomposition. Next, 2:20 or 1:20 (only for H3K4me3) dimensions were used for identifying clusters and for UMAP visualization. For clustering of CoTACIT data from the early 2cell stage, fragment counts in 5 kb genome windows were used for all three histone modifications. The framework for generating a multimodal representation was called the Seurat (v.4) WNN59.
To integrate the six histone modification profiles, we used the multivariate HMM introduced in ChromHMM61. We binarized all .bam files for each synthetic cell using the binarizeBam function of ChromHMM with default parameters. We used the LearnModel function with default parameters to learn 12 states separately on each synthetic cell. To reduce noise and mitochondrial interference, all reads from mitochondrial DNA are filtered out. Next, we annotated each state in three steps, one after the other, to account for the technical noise that may have contributed to the low genome coverage. The twelve states were labelled as a multivalent band of histone modifications, weak promoter, strong promoter, and weak enhancers.
We first created single cell profiles for blastocyst cells and then measured six histone modifications. As described above, we integrated H3K4me1, H3K4me3, H3K27ac and H3K36me3 TACIT profiles as well as H3K27ac CoTACIT profiles with gene expression. We annotated ICM andTE cells based on the expression of their marker genes.
The same procedure was used to integrate H3K 27ac CoTACIT profiles with gene expression. We transfer the H3K 27ac, H3K 27me3 and H3K9me3 profiles to the linked cells.
We ordered scRNA-seq cells along the path and merged five of them into one synthetic cell.
We called peaks for the aggregated.bam files of each histone modification to calculate the genome coverage. We used MACS2 to call peaks with parameters of ‘–nolambda–nomodel -q 0.05–broad’. Next, we binned the mm10 genome into 200-bp genomic intervals, and for each histone modification, genome coverage at a specific stage was calculated as the percentage of genome intervals that overlapped with peaks at that stage. The histone modification signals were defined as covered bins in order to evaluate genome coverage for single cells. The percentage of covered bins was defined as genome coverage for each single cell.
For correlation analysis between different experiments, we calculated the normalized mean scores in 5-kb bins of the genome by using the multiBigwigSummary function in deepTools (v.3.5.1)57. The Spearman correlation or Pearson correlation was calculated between replicates and plotted using the plotCorrelation function.
In vitro superovulation of 2-month-old C57BL/6J female mice using an intraperitoneal injection of 7.5 IU of hCG and PMSG
To obtain pre-implantation embryos, superovulation was induced in 4-week-old C57BL/6J female mice through an intraperitoneal injection of 7.5 international units (IU) of PMSG (San-Sheng Pharmaceutical) followed by 7.5 IU of hCG (San-Sheng Pharmaceutical) 44–48 h later and then the mice were mated with 2-month-old C57BL/6J male mice. Each set of embryos at a specific stage was flushed from oviducts or uteri of pregnant female mice at the following defined time periods after hCG administration: 22–24 h (zygote), 30 h (early 2cell), 43–45 h (late 2cell), 54–56 h (4cell), 68–70 h (8cell), 78–80 h (morula) and 88–90 h (blastocysts). The embryos were kept in M2 medium. The oocytes were collected 48 hours after PMSG administration.
The cells were put in a buffer with a 100 l antibody and then put into the water. At 4 C for 3–4 h, NaCl, 0.5 mM spermidine (Sigma), 2 mM EDTA, 0.01% digitonin, 0.05% TX-100, 1% BSA–PBS, and 10 mM sodium butyrate are present. After incubation, cells were washed twice with 180 μl Dig-wash buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 0.1 mM spermidine, 0.05% digitonin, 0.05% TX-100, 1 cocktail of 10 mM sodium butyrate, and 1 mM PMSF were suspended with a 20 mM high-salt buffer. NaCl, 0.5 mM spermidine (Sigma), 0.01% digitonin, 0.05% TX-100, 1× cocktail, 10 mM sodium butyrate and 1 mM PMSF) containing 3 μg ml–1 PAT–MEA/B. The PAT expression, purification and assembly procedures were performed as per previously described guidelines19. The cells were washed twice with a high salt buffer to remove free PAT from them and then suspended for 10 hours with a cold reaction buffer. Adding 10 l 40 mM stopped the reaction. EDTA and cells were washed twice with 1% BSA–PBS, and single cells were picked and placed into a well of a 96-well plate with a mouth pipette under a microscope. The 96-well plates were pre-rinsed with 1% BSA–PBS to avoid loss of DNA fragments, and 2 μl lysis buffer (10 mM Tris-HCl pH 8.5, 0.05% SDS and 0.1 mg ml–1 proteinase K) was added to each well. Each well contained samples that were covered with 5 l mineral oil and put in a 55 C pot for 15 min to release their genes. Next, 0.5 μl of 10 mM Adding 1 l of 0.9% and the PMSF to each well made a difference in the reaction. Finally, 17 μl PCR mix (0.2 μl Each well was filled with K APA High- GC buffer and 25 mM MgCl2 and 10.8 l H2O. Nextera i5 and 10 mM I7 index primer are the two that are in this table. PCR enrichment was performed in a thermal cycler with the following program: 1 cycle of 72 °C for 5 min; 1 cycle of 95 °C for 3 min; 11 cycles of 98 °C for 20 s, 65 °C for 30 s, 72 °C for 1 min; 1 cycle of 72 °C for 5 min; and hold at 4 °C. The library was selected for Fragments with a percentage of 0.7 and 1AMPure XP beads. The beads are from the AMPure series. The libraries were read with a NovaSeq 6000 platform.
For siRNA knockdown, isolated zygotes were microinjected with sets of three siRNAs against targets (20 μM in total) or with non-target control (NC, 20 μM in total). The following were used for the siRNAs. siRNAs were ordered from Hippobio. The injected embryo’s droplets were cultured in a tissue incubator with mineral oil and then transferred to KSOMaa medium. At the blastocyst stage, embryology was collected, and single-embryoRNA-sette or immunofluorescence staining was used to confirm marker gene expression.
The following antibodies were used for TACIT (catalogue and lot numbers provided after the supplier name): H3K4me1 (1:50; Abcam, ab8895, GR3369516-1); H3K4me3 (1:200; Millipore, 04-745, 3243412); H3K27ac (1:500; Diagenode, C15410196, A1723-0041D); H3K36me3 (1:200; Active Motif, 61101, 06221007); H3K27me3 (1:200; Millipore, 07-449, 3146226); H3K9me3 (1:200; Active Motif, 39161, 30220003); and H2A.Z (1:200, Abcam, ab4174, GR279096-1). Donkey anti-rabbit-Alexa 488 (1:500; Invitrogen, A32790) and donkey anti-rabbit-Alexa 555 (1:500, Invitrogen, A31572) were used as secondary antibodies. Antibodies used in immunofluorescence staining included SOX2 (1:200; Active Motif, 39843, 2226414) and CDX2 (1:200, BioGenex, MU392A-UC, MU392A0516D).
Wild-type V6.5 mouse ES cells were cultured at 37 °C with 5% CO2 and were maintained on 0.1% gelatin-coated plates in high-glucose DMEM culture medium containing 15% fetal bovine serum (Invitrogen), 1% penicillin–streptomycin (Hyclone), 1% MEM nonessential amino acids (Cellgro), 1% Glutamax (Gibco), 1% nucleoside (Millipore), 0.1 mM 2-mercaptoethanol (Sigma) and 1,000 U ml–1 recombinant leukaemia inhibitory factor (Millipore).
Source: Genome-coverage single-cell histone modifications for embryo lineage tracing
Controlled Aging of C57BL/6 Mice at the National Institute of Aging. Data handling and statistical analysis of multiple experiments in M2 medium
A mass of zygotes surrounded by Follar cells was transferred to a single-hyaluronidase solution and held for a few minutes at 37 C. The zygotes were then transferred to M2 medium and their zona pellucida was gently removed by treating with pre-warmed Tyrode’s acidic solution (Sigma) for several minutes. The second polar bodies were removed with a glass needle.
The National Institute on Aging used to have a colony of aging mice. Young C57BL/6 mice were obtained from Jackson Laboratories. Male mice were used in the experiments. mice were kept on a cycle of light and dark, with access to food and water. The procedures and care that were done to the animals comply with the animal welfare act and are approved by the Palo Alto committee on animal research as well as the administrative panel of laboratory animal care.
Data handling and statistical analyses were performed using R studio (v4.1.1) and GraphPad Prism Software unless stated otherwise. All analyses were done using two-tailed Student’s t-tests. All statistical analyses comparing measurements between three or more groups were carried out using one-way ANOVA tests with post hoc tests for multiple comparisons. Significant is the P value of 0.05. To avoid temporal and technical biases, all procedures were carried out in alternating fashion. There is data in Figs. The data figs are 1b–d,h–o, 2b, f–j, 3e–n, 4f–i and 5j–l. 2a, 3f–i, 7a–c,h,i and 8h–k were all successful in two independent experiments.
In fear conditioning tests, mice were trained to associate cage context and an audiovisual cue with an aversive stimulus (foot shock). On day 1 (training), mice were placed in a cage and exposed to two periods of 30 s of paired cue light and 1,000-Hz tone followed by a 2-s foot shock (0.6 mA), with a 180-s interval. On day 2 (contextual fear conditioning), mice were re-exposed to the same cage context and freezing behaviour was recorded during minutes 1.5 to 6 using a FreezeScan tracking system (Cleversys). On day 4 a novel context containing different odors, floor texture, and chamber walls was placed on top of the same cue light and tone from day 1 after 2 min of exploration. Freezing behaviour was recorded for 1.5–6 min following the cue using the FreezeScan tracking system (Cleversys).
The Y maze in the test has 3 white, opaque plastic arms and is 120 from each other. In the beginning of the trials, mice were placed in the end of one arm and allowed to explore the rest of the arms for 5 minutes. All four limbs inside an arm was defined as an arm entry. The maze was cleaned with 70% ethanol between animals and before the first animal to eliminate traces of odour. The number of arm entries and triads (a set of consecutive arm entries) were recorded. The spontaneous alternation was calculated by dividing the number of triads by the number of possible alternations multiplied by 100.
Pseudobulk data normalization and differential gene expression for the determination of cortical and hippocampal nuclei of M. musculus
Pseudobulk counts were derived by aggregating raw counts for each sample using Seurat’s AggregateExpression function. DESeq2 was then used to perform bulk data normalization and differential gene expression across groups using default parameters. P value correction was carried out using the Benjamini–Hochberg procedure (FDR = 0.05) for each comparison. The genes with FDR were used for the enrichment of the KEGG pathway.
Gene counts were obtained by aligning reads to the M. musculus reference genome GRCm38 using CellRanger software (v4.0.0) (10X Genomics). Ambient RNA was removed from each sample using SoupX (v1.6.2) and droplets containing multiple nuclei were filtered out using DoubletFinder (v2.0.4). We used Seurat (v4.1.1) to further exclude cells with fewer than 200 or more than 5,000 features and cells with more than 10% mitochondrial genes. 69,250 nuclei remained and were used for further analysis. 3a–d).
Pooled cortical and hippocampal tissue were dissected from frozen brain hemispheres from young (5-month-old) and aged (19-month-old) mice that had been injected with AAVs 8 weeks prior to takedown. After brain tissue was cut with a razor blade, it was put into glass Dounce tissue grinders and taken off the ice. A 180 twist was included in the 25 strokes used for the homogenization of the tissues. The tissue was put on ice for 5 minutes, and then it wascentrifugationd at a rate of 500g per min. The pellet was resuspended with 4 ml EZ lysis buffer, incubated on ice for another 5 min, and pelleted as before. The pellet was then resuspended with chilled PBS and filtered through a 35-µm strainer before blocking with Mouse Fc block (1:50, BD 553142) in FACS buffer for 5 min. The Nuclei were washed with 0.2 U l1 RN kinases for 30 min and stained with the anti-NeuN product. 50,000 singlet nuclei were sorted using a Sony MA900 Cell Sorter into 1.5m Eppendorf tubes containing 10 and 0.2 U l1 RNase inhibitor. Collected nuclei were centrifuged at 400g for 5 min at 4 °C with slow deceleration. Supernatant was removed leaving about 40 µl of suspended nuclei. Nuclei were counted using a haemocytometer (Sigma, Z359629) and assessed for concentration and quality. snRNA-seq libraries were prepared using the Chromium Single Cell 3′ The kit can be used to target 10,000 nuclei per sample. There are 12 and 11 cycles applied to generate cDNA and the final generation library. High Sensitivity D5000 ScreenTape (Agilent) was used in the quality control of the libraries. The libraries were cataloged on the NovaSeq X.
For production of PHP.V1-sCLDN5::EGFP and all other plasmids containing the PHP.V1 (pUCmini-iCAP-PHP.V1 was a gift from V. Gradinaru; Addgene plasmid #127847) capsid, AAV production was performed in-house utilizing a previously published protocol42. To perform triple transfection of HEK293T cells, the cells were trans cultured on 90–95% confluent cells with Glutamax supplemented and non-essential amino acids. A new warm medium was put in 12 hours after thefection. Medium was collected 72 h post-transfection. Fresh, warm medium was added and collected along with cells 120 h post-transfection and combined with the previous fraction. Cells and medium were centrifuged at 2,000g for 15 min at room temperature. 40% was combined with a separate bottle of supernatant. After 2 hours, the final concentration of 8% wt/vol is put on ice and transferred to 4 C. Cell pellet was resuspended in buffer containing salt-active nuclease (SAN, ArcticZymes Technologies) and incubated at 37 °C for 1 h before transferring to 4 °C overnight. PEG medium was centrifuged at 4,000g for 30 min at 4 °C. The supernatant was thrown away after centrifugation. The pellet was resuspended in the buffer and cultured for 30 minutes at 37 C. Clostyr was loaded onto an iodixanol gradient after being lysated at a room temperature of 2,000g. Gradients were transferred to an ultracentrifuge (Beckman Coulter) using a Type 70 Ti rotor set at 350,000g for 2 h and 25 min at 18 °C. AAV particles were collected from the 40/60% interface, washed inPBS, and concentrated with an Amicon Ultra-15 device with a 100 kDa cutoff. AAV titration was performed using the AAVpro Titration Kit (for Real Time PCR) Ver.2 (Takara Bio). AAVs were injected retroorbitally with 8 to 1011 viral genomes per mouse.
The number is 260 E. Simpson gave the MiniPromoter to him as a gift. A cis rAAV genome plasmid with AAV2 inverted terminal repeats was utilized for cloning of a sCLDN5 and EGFP reporter using restriction enzymes and In-Fusion Snap Assembly (Takara Bio). To knock down C1galt1 in brain endothelial cells, de novo predictions of small interfering RNA (siRNA) guides targeting C1galt1 were generated using the DSIR algorithm39 and subsequently filtered using ‘Sensor rules’ to select for sequences with highly favourable small hairpin RNA (shRNA) features40,41. The three de novo 97-mer miR-E shRNA sequences were synthesised, inserted into pAAV and used for inculturation. A cloned C1GALT1 and B3GNT3 were used to create pA.
For in vitro ROS assays, bEnd.3 cells were plated in 8-well µ-slides (Ibidi, 80826) and treated with 5 nM StcE, 5 μg ml−1 of LPS, or an equivalent volume of saline in complete medium for 16 h at 37 °C. According to manufacturer instructions, cells were washed two times with PBS and cellular ROS assays were done. 20 M of DCFDA and 1 ROS Deep Red Dye were used for their respective tests, and then they were put into a dark room and kept there for 30 minutes. Cells were washed three times and then imaged with a confocal laser- scanning microscope. Quantification of MFI was performed using ImageJ software. For in vivo ROS assays, cerebral microvessel were isolated as described earlier and incubated with 1× ROS Deep Red Dye (Abcam, ab186029) for 30 min at 37 °C in the dark. Microvessels were then washed with 5 ml of PBS and pelleted by centrifugation at 2,000g for 10 min and transferred to 8-well µ-slides (Ibidi, 80826) for immediate imaging on a confocal laser-scanning microscope (Zeiss LSM880). All observed single-plane microvessels were captured and then quantified using ImageJ software.
For confocal imaging analysis, bEnd.3 cells were plated on round coverslips (EMS, 72196-12) in a 24-well plate and treated with 5 nM StcE for 16 h at 37 °C. Cells were fixed in 4% PFC for 15 min, blocked with 3% normal donkey-serum with tictone X-100 in PBS for 1 h at room temperature, and then incubated with the following primary anti-CD31 antibody for 1.5 h. Cells were mounted and coverscovered with Vecta Shield Hardset Antifade Medium with DAPI after being washed three times with PBS. A confocal laser- scanning microscope was used for the scans and images were analysed using ImageJ.
The mouse brain endothelial cell line bEnd.3 (ATCC, CRL-2299) was cultured in high-glucose DMEM (Thermo Fisher Scientific, 10567022) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin and maintained in a humidified incubator containing 5% CO2 at 37 °C. For bulk RNA-seq analysis, bEnd.3 cells were grown in 6-well plates and treated with 5 nM StcE for 16 h at 37 °C. The RNeasy Plus Micro kit was used to lyss cells and collectRNA in Eppendorf tubes. The quantity and quality of genes were assessed by a Bioanalyzer. All samples passed the high quality control threshold and were then taken to the library to be prepared. Libraries were sequenced on the NovaSeq 6000 (paired-end, 2× 150 bp depth). M. musculus has a reference genome. The analysis and visualization of differential expression were done with the DESeq2 (v1.32). Genes with a Padj < 0.05 were used for GO biological pathway enrichment analysis.
Previously published ageing and neurodegenerative disease RNA-seq datasets demonstrating robust brain endothelial cell enrichment were chosen for glycosylation-related gene analysis12,26,27. We searched for related genes that had been listed in the Kyoto Encyclopedia of Genes andTranscripts. The large variety of members in the family meant that most glycoproteins were excluded. Significant enrichment was achieved with Padj 0.05 set as the threshold for significant enrichment, in order to analyse upregulated and downregulated genes.
LC–MS/MS analysis was performed on a Q Exactive HF-X (Thermo Fisher Scientific) with an UltiMate 3000 RSLCnano system (Thermo Fisher Scientific). Peptides were loaded on an in-house 75-μm (inner diameter) capillary column packed with 40 cm of ReproSil-Pur 120 C18-AQ 1.9 μm resin (Dr. Maisch). Chromatographic separation was achieved using a flow rate of 300 nl min−1 with the following 120 min gradient: 96% A + 4% B for 18 min, 70% A + 30% B for 72 min, 60% A + 40% B for 15 min, and 4% A + 96% B for 15 min, where solvent A was 0.1 % formic acid in HPLC-grade water (Fisher) and solvent B was 0.1% formic acid in HPLC-grade acetonitrile (Fisher). The scanned material was obtained at a resolution of 60,000, with an automatic gain control (AGC) target of 3 106, and a maximum injection time of 20 ms. MS2 scans were acquired with the following parameters: resolution of 15,000, AGC target of 1 × 105, maximum IT of 54 ms, loop count 15, TopN 15, isolation window 1.4 m/z, fixed first mass 100.0 m/z, normalized collision energy (NCE) 28 units, charge exclusion of unassigned, 1, 6–8 and >8, peptide match preferred, exclude isotopes on, and fragmented m/z values were dynamically excluded from further selection for a period of 45 s. Raw data were processed and analysed using MaxQuant and Perseus36. In brief, peptide spectral matches were made against a target-decoy Mus musculus reference proteome database downloaded from Uniprot. Methionine oxidation and N-terminal acetylation were specified as variable modifications, and carbamidomethylation of cysteines was specified as a fixed modification. For searches, the pre-cursor and product ion mass tolerances were used. The unique and razor peptides were used. The results were taken to a 1% false discovery rate. The minimum ratio count was set to 1 for the MaxLFQ 37 used to calculate the levels of the expressed genes. For quantitative comparative analysis, protein intensity values were log2-transformed, and missing values were imputed from a normal distribution with width 0.3 and downshift value of 1.8 using Perseus. The PCA was performed in Perseus using the Benjamini–Hochberg FDR. The Go term enrichments used DAVID38 to background the M. musculus proteome.
The Human Immune Monitoring Center at the Cardinal University has a fluorescent reagent that can be used to measureplasma cytokine levels. The instructions for the mouse were provided by the manufacturer. Plasma samples were diluted 1:3 and run in singlet on a 96-well plate alongside standard curve and quality control samples. Custom Assay Chex control beads (Radix BioSolutions) were added to all wells to assess nonspecific binding.
Source: Glycocalyx dysregulation impairs blood–brain barrier in ageing and disease
Endothelial Glycocalyx Layer in Mice: Treatment by Structural Injection with a 1-mm-thick Fixation Matrix
Transmission electron micrographs of the brain endothelial glycocalyx layer were obtained as described4 with some modifications. The young mice were perfused with the ice-cold fixation buffer composed of 2%glutaraldehyde (EMS), 2% sucrose, 1%sodium cacodylate buffer, and 2% lanthanum nitrate. The brain was removed and sliced coronally using a matrix into 1-mm-thick sections. After being immersed in a perfusion solution for 2 h,tical punches were cut and put into a solution that did not containglutaraldehyde. The samples were washed with a 0.03 M NaOH 2% sucrose solution and then subjected to an upwelling of the ethanol and being embedded in the EMS. 90 nm sections were cut using a Leica UC6 ultramicrotome (Leica Microsystems) and collected onto formvar-coated 50-mesh copper grids. The grids were post-stained with 2% uranyl acetate followed by Reynold’s lead citrate for 5 min each. Sections were imaged using a Tecnai 12 120 kV TEM (FEI), and data were recorded using either an UltraScan 1000 with Digital Micrograph 3 software (Gatan) or a Rio16 CMOS camera with GWS software (Gatan). The animals were captured by a blinded observer with between 6 and 12 cortical capillaries. Quantitative analysis of endothelial glycocalyx thickness and area were performed using ImageJ software (Extended Data Fig. 1b). For StcE treatment experiments, 3-month-old mice were retro-orbitally injected with 0.25 mg kg−1 StcE or inactivated StcE(E447D) 24 h before perfusion.
Mice were anaesthetized and injected retro-orbitally with Sulfo-NHS-biotin (Thermo Fisher Scientific, 21335) at 0.25 mg g−1 body weight. After 5 minutes of circulating the tracer was able to be taken out and put in PBS. Hemibrains were post-fixed in 4% PFA overnight at 4 °C, cryopreserved in 30% sucrose, and sagittally sectioned into 40-μm slices. Sections were co-stained with CD31 and the appropriate secondary Immunoglobulin as described, and they were also blocked. Images were taken on a confocal laser-scanning microscope and analysed using ImageJ software. Multicoloured gradient images were generated using the fire LUT in ImageJ. Permeability index of vessels was determined as the area occupied by tracer divided by the vessel area.
The mice were euthanized with 2.5%. The following ice-cold solutions can be used for avertin and transcardially perfused via the peristaltic pump. The mice were euthanized with 2.5% of their body weight. Unless noted otherwise, avertin and manually perfused with PBS. The tissues were fixed in 4% PFA at 4C overnight and then preserved in 30% PBS. Tissues were sectioned into 40 μm slices using a microtome (Leica). At room temperature and 4C overnight, a goat anti-CD31 (1: 100, R&D, asf 3628) was added to the normal donkey serum and slices were blocked. The following day, slices were washed three times with TBS-T, stained with the appropriate Alexa Fluor-conjugated secondary antibodies (1:250, Thermo Fisher Scientific) or Alexa Fluor-conjugated streptavidin (1:1,000, Thermo Fisher Scientific) for 2 h at room temperature, washed three times again, mounted, and coverslipped with Vectashield Hardset Antifade Mounting Medium with DAPI (Vector Labs, H-1500-10). Imaging was performed on a confocal laser-scanning microscope (Zeiss LSM880), and images were analysed using ImageJ. Luminal vascular coverage was calculated as vessel (CD31+ or COL4A+) area occupied by the marker of interest divided by total vessel area. The MFI was calculated using CD31+ mask.
The brain tissues were obtained from the Clinical Research Center with patient consent and approval from the Institutional Review Board. There was no need for autopsies to take more than 12 h after death because the samples were kept in a high temperature until the time of processing. The information is summarized in Supplementary Data 2. Both clinically and pathologicalally, individuals in the Alzheimer’s disease group exhibit Alzheimer’s brain disease hallmarks.
In PBS, the mice were injected with a small amount of HRP type II in order to perform the ultrastructural analysis. After 30 min, the brain was fixed with a 0.1 Msodium cacodylate buffer and 4% to 5% of glutaraldehyde and 4% PFC at a room temperature. The tissues were washed with 0.1 M sodium cacodylate before they were sliced using a matrix of 1-mm-thick sections. Thetical punches were cut and placed in a container with.05% hydrogen peroxide and 3,3′-diaminobenzidine for 45 min at a room temperature. There was a stain on the tissue after it was washed with TBS and after it was fixed with 2% osmium tetroxide and 2.5% potassium ferrocyanide. Samples were then dehydrated in an ascending ethanol gradient and embedded in epoxy resin (EMS). The section was cut using the UC7 ultramicrotome and collected on the formvar-coated copper grids. The grids were post-stained with 3.5% uranyl acetate followed by Sato’s lead citrate. Sections were imaged using a Tecnai 12 120 kV TEM (FEI), and data were recorded using a Rio16 CMOS camera with GWS software (Gatan). The assessment of tight junctions in the images was performed in a blinded manner.
A new study has found that genome-coverage single-cell histone modifications could be used to tracing lineages in embryos. Researchers used fragment counts in 5kb genome windows to assess genome coverage for all three of the histone modifications. This was done using data from a CoTACIT database of embryo lineages from pregnant female mice.