A human atlas has a continuous tissue axis
Annotation of the CITE-seq data on log-normalized RNA and denoised ADT modalities using WNN and sPCA
Annotation of the CITE-seq data was carried out on integrated RNA and denoised ADT modalities. For this purpose, both data modalities were log-normalized and the top 2,000 highly variable genes were identified, followed by PCA on the scaled highly variable genes using the standard functions in the Seurat package. MNN correction was applied to the PCA loadings matrix using the reducedMNN function from the Batchelor package60 (v.1.18.1) to reduce batch and donor effects between samples. To integrate both modalities, multimodal neighbours and modality weights were identified and a weighted nearest neighbours (WNN) graph58 was constructed using FindMultimodalNeighbors based on the PCA. The number of PCs to be used was decided by the variation of consecutive PCs. A UMAP visualization was created using the WNN graph to show the combination of the two therapies. sPCA was performed on transcriptome data using the RunSPCA function of the Seurat package to obtain a reduction in the number ofRNAs that represent the structure of the WNN graph58
Drug and target gene information for humans (Homo sapiens) were gathered from the ChEMBL database (https://www.ebi.ac.uk/chembl/). We searched the clinically approved drugs that target genes and found 65 of them that carried warnings of teratogenicity (Supplementary Table 6). The drug scores were calculated. Drug categories were introduced based on the broad clinical utility. The Drug2Cell Python package can be found at GitHub.
Ligand–receptor interactions were inferred using ‘cpdb_analysis_method.call’ in CellPhoneDB (v4.0.0). The genes that were expressed in more than 10% of cells within each cluster were included. Inferred interactions with a P > 0.001 were removed. We used NicheNet (v1.1.1) to identify different interactions between endochondral and intramembranous niches. The minimum log fold change per cluster was used in the Wilcoxon test to calculate DEGs of osteogenic clusters and tip cells across the two niches. The calculation of the ligand activities were done using the top 1,000 DEGs. We prioritized the ligand–receptor links using default settings. The top ten ligands were visualized using heatmaps.
We used a network propagation98 approach to integrate GWAS summary statistics and cell cluster marker gene-based scores for prioritizing disease-relevant and cell cluster-specific subunits of our transcription factor network. First, scores per SNP were computed from downloaded summary statistics and weighted by linkage disequilibrium. Then, the scores were mapped to a GRN, which is an enhancer-driven GRN that is computed with SCENIC+. As the networks contain transcription factors and target genes, and also regions with transcription factor-binding sites as nodes, the SNP scores were mapped to both genes and regions. The scores were replicated across the network using a random walk with restart process. This integrates the contribution of individual SNPs, with signals converging around relevant network nodes. The procedure was repeated with 1,000 randomly permuted scores to compute permutation-test results and z-scores. Next, differential expression-based marker gene scores for each cell cluster were propagated in the same way, resulting in cell cluster specificity scores for each network node. The SNP and expression-based scores were then combined per cell cluster (as in ref. 99) By using the minimum for each step. The final scores were thresholded, and the resulting connected components were obtained as enriched subnetworks. The method can be found in a tool called SNP2Cell, which is accessible at http://www.Teiclab.com.
The analysis 97 has been applied to identify cell clusters that are related to the disease. The model makes use of full summary statistics from GWAS, linking single-nucleotide polymorphisms (SNPs) to genes, and captures a general trend between gene expression and disease association of linked loci for each cell cluster. The model corrects for linkage disequilibrium and other factors. We used the full GWAS summary statistics obtained from the catalog, as well as total knee and hip replacement. 62 (https://msk.hugeamp.org/downloads.html; Supplementary Table 8).
In the osteogenic subcompartment, non-cycling droplets were subsetted and X_scVI was used as projections for palantir. A neighbourhood graph was generated on the diffusion space using sc.pp.neighbors, and the first two principal components were used as initial positions to create ForceAtlas2 embeddings using sc.tl.draw_graph. scFates85 (v1.0.3) was used to predict a principle graph that captures the differentiation path. The force-directed embeddings and principle graph were exported into R, and monocle3 (v1.0.0)86,87 was used to compute differentially expressed genes along pseudotime using a graph-based test (morans’ I)87,88, which allows identification of genes upregulated at any point in pseudotime. The results were visualized using heatmaps with smoothing with smoothing splines in R. Velocity analysis91 was performed using scvelo92 (v0.2.3). The read counts were computed with velocyto from the data before using scvelo.pp.moments, scvelo.tl. velocity and scvelo.tl.velocity_graph. CellsRank94 was used to obtain an independent prediction of directionality by the assumption that the number of expressed genes decreases during differentiation.
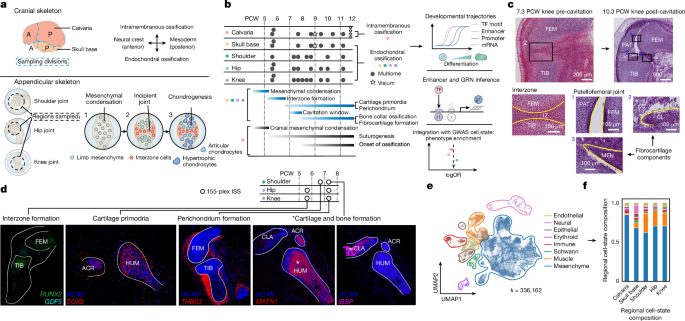
To approximate the timing of the start of cavitation, we used a specific gene set within the hip, shoulder and knee joints. We excluded genes with low expression levels in our data, such as HAS3. For pathway analysis, we retrieved the Gene Ontology terms and computed the correlation between their enrichment scores and cavitation enrichment scores.
OrganAxis and ISS-Patcher: an extrapolation method for tissue annotations and application to missing scRNA genes
OrganAxis attempts to derive a relative, signed position of a point in space in relation to two landmarks. The OrganAxis base function H is highly flexible and tuneable with respect to the research question, spatial resolution and sampling frequency. A guide to tissue annotations is provided in Supplementary Notes 1 and 2. The CMA is an extrapolation of OrganAxis, which is defined by a weighted linear combination of two H functions (CMA = 0.2 × H(edge-to-cortex) + 0.8 × H(cortex-to-medulla)). All IBEX and Visium images were annotated with TissueTag v.0.1.1 at a resolution of 2 um per pixel (ppm = 0.5). Then, annotations were transferred to a quasi-hexagonal grid that was generated by placing points with r-microns spacing in the x and y directions and staggering every other row by r/2. r is the number we used throughout the study. L2 distances to broad level annotations (annotation_level_0) and the corresponding CMA values were calculated with k = 10 for all spots in the hexagonal grid. The distances to fine annotations for L2 were calculated with k numbers and transferred to spots in Visium or IBEX.
ISS-Patcher is a package for approximating features not experimentally captured in low-dimensional data based on related high-dimensional data. The approach was developed using snRNAseq data as a reference in the study, and was used to approximate expression signatures for missing genes. The 155–158 genes present in the dart pool was sub-coded and then adjusted to median total cell counts, log-transformation and z-scoring for the two datasets. The 15 nearest neighbours were identified by using the Annoy Python package, and the genes missing from the International Space Station were also imputed as average raw counts of scRNA-seq neighbours.
We performed Cell2location (v0.1.4) for deconvolution of Visium CytAssist voxels using our annotated Multiome data as inputs. Sample donor was used as the batch variable, and each library was considered a covariate in the regression model. For spatial mapping, we estimated 30 cells per voxel based on histological data, and set a hyperparameter detection alpha value of 20 for per-voxel normalization.
We applied the Python implementation of the MILO package (v0.1.1) for differential abundance testing (http://github.com/emdann/milopy)82. We used the scVI latent representation to create a k-nearest neighbour graph of droplets in the relevant compartment and subsequently applied milopy to allocate droplets to overlapping neighbourhoods, with these droplets originating from multiple samples (brc_code). Each neighbourhood was categorized based on the votes from the majority of the population. The values of the anterior–posterior position and the calvarium-appendicular co predictors were binarized to allow testing. By using the Bejanmini–Hochberg correction, we determined log fold-change values for differential abundance and false discovery rate values.
For the focused multimodal analysis, we subsetted the full scRNA-seq dataset to include only paediatric data and further removed T lineage cells without CITE-seq protein information. We then performed cell2location as described above to obtain deconvolved cell type mapping based on CITE-seq annotations. In addition, as cell2location deconvolves spots by unique pseudobulk expression profiles from a single-cell reference based on discrete cell annotations, the mapping resolution is limited to that of the annotated cell subsets. The clustering of CD4/CD8 cell groups using high-resolution scans was used to investigate the continuous spatio temporal nature of the cells. These cell clusters were then mapped to our paediatric Visium data with cell2location as described above. To measure the position of each cell cluster with the highest density labels we used the weighted mean to find Visium spots with the lowest density. The cells consisting of the associated cluster were assigned the value. Further details are provided in the GitHub repository for this Article (see Code availability).
Feature barcode (FB), gene expression (GEX) and TCR libraries were prepared according to protocol CG000330 Rev A (10x Genomics) using the Chromium Next GEM Single Cell 5′GEM The Library Construction Kit (10x Genomics, 1000190), 5′ Feature barcode kit (10x Genomics, 1000128), Human TCR Amplification Kit (10x Genomics, 1000252), and dual index kit are all included in the kit. The protocol version for >6,000 cells was followed and libraries were amplified for 13 cycles (cDNA), 14 cycles (GEX), 8 cycles (FB) or 12 + 10 cycles (TCR libraries). Library quality and quantity were checked on the Bioanalyzer instrument (Agilent) using a high-sensitivity DNA assay. Libraries were pooled and sequenced on the NovaSeq 6000 instrument (Illumina) to a minimum of 25,000 reads per cell for GEX, 10,000 reads per cell for FB and 5,000 reads per cell for TCR libraries.
The manufacturer’s protocol led to the performing of Visium CytAssist Spatial Gene Expression for Fresh Frozen. Regions of interest were selected based on the presence of microenvironments of bone formation relevant to the droplet data (for example, coronal suture) and aligned to the CytAssist machine gasket accordingly. Images were captured using a Hamamatsu S60 slide scanner at ×40 magnification before conducting the Visium CytAssist protocol for subsequent alignment. SpaceRanger was used to map the libraries.
Li et al.65 outlines the decoding method we used. This pipeline consists of five distinct steps. First, we performed image stitching using Acapella scripts provided by Perkin Elmer, which generated two-dimensional maximum intensity projections of all channels for each cycle. Next, we used Microaligner66 (v1.0.0) to register all cycles based on DAPI signals using the default parameters. For cell segmentation, we utilized a scalable algorithm that leverages CellPose67 (v3.0) as the segmentation method. The cell size has been set to 70 pixels in diameter and 10pixels in total to mimic the cytoplasm. We used the PoST code68 (v1.0) with the following parameters: rnaspot_size, trackpy_percentile, and trackpy_separation. We generated AnnData objects from the data we gave to the cells, and then assigned the decoded RNA molecule to the cells. Cells with more than fourRNAs were the ones kept for analysis.
Automation of Whole-Cell Excavation and Imaging of Donor Tissue using a RNAscope with Multiplex Fluorescent Reagent Kit v2
We applied whole-cell dissociation of fresh donor tissue as previously described5. Before cell excavation, the sample tissues were dissected to obtain bone and soft tissue samples. The cell suspension was stained with idevitrogen for live viability, FGFR3 antibody, and TACR3 antibodies. Singlet cells that were DAPI-positive were gated for DAPI staining with the help of a Big Foot cell sorter and its proprietary software. Sequential gating for FGFR3 and TACR3 was then conducted to identify double-positive cells. Positive and negative controls for FGFR3 and TACR3 were conducted with the help of human peripheral blood mononuclear cells.
Cryosections were processed using a Leica BOND RX to automate staining with the RNAscope Multiplex Fluorescent Reagent Kit v2 assay (Advanced Cell Diagnostics and Bio-Techne), according to the manufacturers’ instructions. Supplementary Table 7 contains probes. Before staining, fresh frozen sections were post-fixed in 4% paraformaldehyde in PBS for 45 min at 4 °C, then dehydrated through a series of 50%, 70%, 100% and 100% ethanol for 5 min each. Following manual pre-treatment, automated processing included digestion with Protease III for 15 min before probe hybridization. Tyramide signal amplification with Opal 520, Opal 570 and Opal 650 (Akoya Biosciences), TSA-biotin (TSA Plus Biotin Kit, Perkin Elmer) and streptavidin-conjugated Atto 425 (Sigma-Aldrich) was used to develop RNAscope probe channels. The stained sections are similar to those in the above picture.
The samples64 were cleared using the iDISCO+ protocol. The samples were placed in the tubes under the rotating agitation and dehydrated for 1 h. The samples volume was about five times that of the Methanol volumes. The samples were next incubated in a solution of 67% DCM and 33% MeOH overnight followed by 100% DCM for 30 min at room temperature on a rotator, then put in 100% DBE. The cleared samples were imaged with a light-sheet microscope which has a sCMOS camera and is controlled by Imspector Pro. The four different wavelength lasers generated a light sheet which was 4 m in thickness. One or more objectives had different magnifications of 0.6, 1, and 1.66 The samples were supported by a sample holder from miltenyi and placed in a tank filled with DBE and illuminated with a laser light sheet from one or both sides. LightSpeed Mode was used to acquire the images in a reasonable amount of time. There were mosaics assembled with an overlap of 10%. The images were acquired in a 16 bits TIFF format. Automatic alignment of the tiles was performed after the images were processed using MACS iQ View Software. Stack images were converted to imaris file (.ims) using ImarisFileConverter. The surface tool was used to get at the specific structure in Imaris, which was then masked with the surface. Images and videos were taken by using either the function Snapshot and Animation in Imaris. Adobe Photoshop (v25.2) was used to colour the suture areas.
Specimens were cultured for a week in EDTA at room temperature. The solution had been renewed halfway through the period. The samples were washed twice in PBS 1X during 1 day. Samples were dehydrated for 1 h at room temperature in ascending concentrations of methanol in H2O (20%, 40%, 60% and 80%). Then, samples were placed overnight under white light (11 W and 3,000 K°) and rolling agitation (004011000, IKA) with a 6% hydrogen peroxide solution in 100% methanol. Samples were rehydrated for 1 h at room temperature in descending concentrations of methanol (80%, 60%, 40% and 20%), washed twice and blocked in 0.2% PBS-gelatin with 0.5% Triton X-100 (PBSGT) solution during 1 week. The samples were put in a solution with the primary antibodies and put into a tank at 37 C for 14 days. This was followed by six washes of 1 h in PBSGT at room temperature. Next, secondary antibodies were diluted in PBSGT and passed through a 0.22-μm filter. After 7 days of sterilization at 37 C, the samples were washed six time in PBSGT at room temperature.
ISS was performed using the 10X Genomics CARTANA HS Library Preparation Kit (1110-02, following protocol D025) and the In Situ Sequencing Kit (3110-02, following protocol D100), which comprise a commercialized version of HybRISS63. Probe panel design was based on fold-change thresholds in cell states of the limbs (Supplementary Table 3). In brief, cryosections of developing limbs were fixed in 3.7% formaldehyde (252549, Merck) in PBS for 30 min and washed twice in PBS for 1 min each before permeabilization. Sections were briefly digested with 0.5 mg ml−1 pepsin (P7012, Merck) in 0.1 M HCl (10325710, Fisher) at 37 °C for 15 s (5 PCW) or 30 s (6 PCW and older), then washed twice again in PBS, all at room temperature. Following dehydration in 70% and 100%, the chamber was attached to each slide with 50 l volume. Rehydration in buffer WB3 led to probe hybridization taking 16 hours at 37C. 5 padlock probes per genes were included in the 158-plex probe panel. The section was washed with PBS-T (PBS with 0.05% Tween-20) twice, then with buffer WB4 for 30 min at 37 °C, and three times again with PBS-T. Probe ligation in RM2 was conducted for 2 h at 37 °C, and the section was washed three times with PBS-T, then rolling circle amplification in RM3 was conducted for 18 h at 30 °C. Following PBS-T washes, all rolling circle products (RCPs) were hybridized with LM (Cy5-labelling mix with DAPI) for 30 min at room temperature, the section was washed with PBS-T and dehydrated with 70% and 100% ethanol. The hybridization chamber was removed and the slide mounted with SlowFade Gold Antifade Mountant (S36937, Thermo). Quality control is a step that has been taken to identify spots during decoding and confirmation of Cy5-labeling RCPs. The 64 (NA 1.15) water-immersion objective was used to conduct the film test using the Perkin Opera Phenix Plus High-Content Screening System. For channels: DAPI (excitation of 375 nm and emission of 480 nm), Atto 327 (excitation of 335 nanometers and emission of 355 nanometers), alexa ferri 488 (excitation of 478 nanometers and emission of 475 nanometers), After the procedure, all slides were put in PBS, with minimal movement, so that the coverslip wouldn’t fall off. The section was dehydrated with 70% and 100% ethanol, and a new hybridization chamber was secured to the slide. The previous cycle was stripped using 100% formamide (AM9342, Thermo), which was applied fresh each minute for 5 min, then washed with PBS-T. Barcode labelling was conducted using two rounds of hybridization, first an adapter probe pool (AP mixes AP1-AP6, in subsequent cycles), then a sequencing pool (SP mix, customized with Atto 425), each for 1 h at 37 °C with PBS-T washes in between and after. The section was dehydrated, the chamber removed, and the slide mounted and imaged as previously described. This was repeated another five times to generate the full dataset of seven cycles (anchor and six barcode bits).
All sample donors were granted written and informed consent for the use of the developing human limb and cranium tissue samples, with full approval from the Cambridge Central Research Ethics Committee. After dissection the samples were frozen in PBS at 4C. Shoulder, hip and knee joints were dissected en-bloc from the limbs. For the shoulder joint, a proximal incision was made at the distal third of the clavicle, and a distal incision was created at the neck of the humerus. The glenohumeral and the acromioclavicular joints were captured in approximations when there were no distinctive bone features. For the cranium samples (less than 8 PCW), two regions were dissected for each of the calvaria and skull base, separated at the posterior border of the frontal bone in both cases. For older cranial samples, tissues were sliced apart to separate the bones where possible. Samples were initially embedded in optimal cutting temperature medium and frozen at −80 °C on an isopentane-dry ice slurry. Using a cryostat was used to cut the sections using 10 m thickness and placed onto SuperFrost Plus slides for Visium CytAssist or single-nucleus processing. The consent of all of the sample donors is obtained before samples can be used in whole mount immunostaining. The HuDeCA Biobank gave samples and they were used in accordance with French regulations. Full authorization to use these tissues was granted by the French agency for biomedical research (Agence de la Biomédecine, Saint-Denis La Plaine, France; PFS19-012) and the INSERM Ethics Committee (IRB00003888).
TCR-sequencing data and milo neighbourhoods in a human thymus cell atlas mapped to a continuous tissue axis
To make a trajectory inference for T cells, CSITE-seq data had to be subsetted. A new WNN UMAP was made using the surface Protein andRNA. Slingshot62 (v.2.6.0) was used to establish a minimum spanning tree on the WNN UMAP using the getLineages function based on mutual nearest neighbour-based distance with DP_pos_sel set as start point and SP_CD4_mature and SP_CD8_mature specified as end points. The derived pseudotime orderings were used to assess transcript and surface marker expression throughout differentiation, which was obtained using the get curves function.
A notebook can be found in the repository for this article, which contains single-cell TCR-sequencing data. All the combined libraries constructed a Dandelion class object. The cells were then further subset to only include the S and P cells that contained V and J rearrangements. Next, milo neighbourhoods were constructed based on scVI neighbourhood graphs (n_neighbors = 100) and VDJ genes and frequency feature space was calculated.
The data was processed in the R packages Seurat58, SeuratObject, SingleCellExperiment, and Matrix. Data were visualized using ggplot2 (v.3.5.0), ggrastr (v.1.0.2), ggridges (v.0.5.6) and RColorBrewer (v.1.1-3).
Source: A spatial human thymus cell atlas mapped to a continuous tissue axis
IBEX thymus antibody panel for imaging and staining with frozen sections using a PELCO Thermoelectric Recirculating Chirp Cooler
IBEX imaging was performed on fixed frozen sections as described previously17,53. The 20 m sections were cut on a cryostat and then adhere to two well chambered cover glasses with 15 l of chrome alum Geranium per well. The section that was frozen was permeabilization, blocked and stained in PBS with a 1% bovine albumin and 1% human Fc block. The PELCO was used for this procedure. The microwave equipped with a PELCO SteadyTemp is called the BioWave Pro 36500-230. The Pro 50062 thermoelectric recirculating chiller is powered by a 2-1-2-1-2-1-2 program. The IBEX thymus antibody panel can be found in Supplementary Table 3 and has been formatted as an Organ Mapping Antibody Panel54 (OMAP-17) accessible online (https://humanatlas.io/omap). Custom antibodies were purchased from BioLegend or conjugated in house using labelling kits for Lumican (AAT Bioquest, 1230) and LYVE-1 (Thermo Fisher Scientific, A20182). A biotin avidin kit (Abcam, ab64212) was used to block endogenous avidin, biotin and biotin-binding proteins before streptavidin application. Cell nuclei were visualized with Hoechst 33342 (Biotium) and the sections were mounted using Fluoromount G (Southern Biotech). After image acquisition and before the removal of the mounting medium, PBS was used. After each staining and imaging cycle, the samples were treated with two 15 min treatments of 1 mg ml−1 of LiBH4 (STREM Chemicals) prepared in deionized H2O to bleach all fluorophores except Hoechst.
Human thymus samples were obtained from the pathology department of the Children’s National Medical Center in Washington, DC, after cardiothoracic surgery from children with congenital heart disease, as thymic tissue is routinely removed and discarded to gain adequate exposure of the retrosternal operative field. The use of the thymus samples for this study was exempt from review due to guidelines issued by the Office of Human Research Protections. There were no genetic concerns for the patients in this cohort. There are details about the cohort in Supplementary Table 2. Human thymi were placed in PBS on receipt and processed within 24 h after surgery. Excess fat and connective tissue were trimmed and sectioned into <5 mm cubes. For IBEX image analysis, human thymi were fixed with a mixture of trypsin and cytoperm. After fixation, all tissues were washed for 5 minutes in PBS and then put in a 30% sugar solution for 2 days before being put in the OCT compound.
Source: A spatial human thymus cell atlas mapped to a continuous tissue axis
The Perkin Elmer Operetta CLS High Content Analysis System (ISS Patcher KNN Mapping) applied to RNA/Protein Sequences and to T Cells
There are contrast used for the extended data fig. The following were the following: LY75, IGFBP6, LAP, and red 300–2500.
Confocal imaging was performed on the Perkin Elmer Operetta CLS High Content Analysis System using a ×20 (NA 0.16, 0.299 μm px−1) water-immersion objective with 9-11 z-stacks with 2 µm step. Channels: DAPI (excitation, 355–385 nm; emission, 430–500 nm), Atto 425 (excitation, 435–460 nm; emission, 470–515 nm), Opal 520 (excitation, 460–490 nm; emission, 500–550 nm), Opal 570 (excitation, 530–560 nm; emission, 570–620 nm), Opal 650 (excitation, 615–645 nm; emission, 655–760 nm). The individual image stacks were visualized using a proprietary A capella script provided by Perkin Elmer. Plus (Glencoe Software).
We used the KNN program to identify T cell types in the IBEX data, and applied the patcher to the Cite-seq T lineage data. We then used the CITE-seq data as a reference to repeat the KNN-based annotation on these selected cells. This KNN-based reannotation was performed on a hybrid RNA/protein reference, which included protein measurements for the 19 markers assayed in both CITE-seq and IBEX in addition to RNA measurements for the remaining 23 genes as described in Supplementary Table 11. We used the same KNN implementation as described above but with k = 7, while also imputing CD4 and CD8 pseudotime.
The general code for the ISS patcher KNN mapping algorithm can be found in the dedicated GitHub repository (https://github.com/Teichlab/iss_patcher/tree/main) and the full example for KNN mapping using IBEX is reported in the GitHub repository for this Article (see Code availability).
Source: A spatial human thymus cell atlas mapped to a continuous tissue axis
Extraction and analysis of IBEX single-cell annotations with a large number of non-specific and non-non-specific cells in the medulla
In some of our IBEX samples AIRE staining in particular produced a relatively high level of non-specific signal and low signal-to-noise ratio. AIRE+ mTECII cells are predicted to not be in the medulla and we identify some of the ones that are. We had cases of individual samples for which a specific antibody did not perform well or was missing (for example, for IBEX_01 the KRT10 antibody was out of stock). However, as ISS patcher was run on each sample separately, this did not affect proper scaling of that marker and its use of it for mapping in other samples. Moreover, by selecting cells with a higher proportion of matched KNN cells from the single-cell data (KKNf) these effects are reconciled through the removal of cells with low-confidence mapping. We would like to flag this point for future researchers who want to reuse our datasets.
On the basis of majority voting, the IBEX query cell was given the most frequent cell annotations in the GEX reference. The evidence fraction is the percentage of KNNs that contributed to the majority voting. The IBEX cell received the label A if out of 30 nearest neighbours, 13 were labeled as A, 10 as B and 7 as C.
The KNN of the low-dimensional observations (IBEX) in the high-dimensional space (GEX) were identified. The counts of absent features were imputed as the mean of the high-dimensional neighbours.
All sample single-cell segments were merged into a single AnnData object totalling more than one million cells.
Signal intensity extraction: for each cell mask post-filtering, we extracted the mean and maximum signal intensity for each channel in the IBEX image. The cellXprotein file was produced for each sample.
Handling high nuclear density: the high nuclear density, especially in the thymic cortex, produced scenarios in which segmentation masks were often bordered by neighbouring cells, resulting in substantial inter-cell signal bleed. The interface between the cells was excluded because the mask boundary was not protected if no other cell was present.
Segmentation: the image arrays were sequentially segmented with cellpose using specific parameters, for example, diameter, resampling, anisotropy, thresholds and batch size. We used tiles to overcome restrictions on GPU and RAM resources.
The individual image were separated by plane and the sample channel’s metadata was made into a.csv file. The nuclear staining channel (Hoechst) was located, and a set of tiled 3D image arrays was generated.
The representative sections were acquired with a confocal microscope that had a 40 objective, 4 HyD and 1 PMT detectors, a white-light laser as well as a red- light source. Panels consisted of antibodies conjugated to the following fluorophores and dyes: Hoechst, Alexa Fluor (AF)488, FITC, AF532, phycoerythrin (PE), eF570, AF555, iFluor 594, AF647, eF660, AF680 and AF700. All images were captured at an 8-bit depth, with a line average of 3 and 1024 × 1024 format with the following pixel dimensions: x (0.284 μm), y (0.284 μm) and z (1 μm). The images were tiled and merged using the LAS X Navigator software. When selecting a tissue section for IBEX, multiple sections of tissue were examined before choosing a section that contained several distinct parts, which resulted in an unusual region of interest. The emission was collected on different detectors with different types of fluorophores used to minimize spillover. The Channel Dye Separation module was used to correct residual spillover in LAS X. For publication-quality images, Gaussian filters, brightness/contrast adjustments and channel masks were applied uniformly to all images. SimpleITK55,56 was used to perform image alignment of IBEX panels. Additional details on antibodies, protocols and software can be found on the IBEX Knowledge-Base (https://doi.org/10.5281/zenodo.7693279).
To identify genes exclusively expressed in a specific cell type or subset thereof (‘specialization genes’, SGs), we developed custom Python functions. From the read count, the expression of the genes was scaled. Cells that showed expression below a cut-off of 0.05 and genes that had expression below 1.5 mean counts were excluded from further steps. A quantile threshold was used to select cells with the highest expression levels. A test to determine if the cells were overrepresented in a specific cell type was done for each of the genes. The genes that were not expected to be expressed in more than one cell type were considered to be segregating.
To estimate the average position of a cell or gene distribution along CMA and HC axes (L2 distance to the nearest HC), spots with low gene expression were filtered out by using appropriate thresholds (0.2 for scVI corrected gene expression and 0.5 for predicted cell abundances). The position of a gene or cell was then calculated according to the following formula: for every gene/cell and axis positions, the weighted mean was calculated as a dot product of spot cell abundance values and CMA position divided by the sum of the cell abundance values across spots.
We mapped the fetal and paediatric datasets using cell2location51 v. 0.1.3 separately. We therefore subset our single-cell reference datasets according to either fetal or paediatric stage and removed rare cell types that were predominantly found in one of these stages. We removed cell types that showed stress signatures because we believed they came from technical factors. A list of cell types that were excluded and the exclusion criteria are provided in Supplementary Table 9. In order to deconvolution, we removed cell cycle genes and other genes using the expression TR[AB][VDJ]$IG[HKL]VDJC’. We then further calculated highly variable genes and used relevant metadata cofactors (sample, chemistry, study, donor, age) to correct for batch effects in the cell2location model.
Before performing any association analysis with the CMA, we further removed lobules (based on ‘annotations_lobules_0’) that had no or small medullar or cortical regions, as we expected our CMA model to be less accurate in these cases.
We subsequently used TissueTag v.0.1.1 for semiautomated image annotation (Supplementary Note 1). Cortical and medullary pixels were predicted with a pixel random forest classifier by generating training annotations based on spots with the highest gene expression of AIRE (for medulla) and ARPP21 (for cortex). Automatic cortex/medulla annotations were then adjusted manually where necessary. We annotated individual thymic lobes, as well as structures including capsule/edge, freezing/sectioning artefacts, HCs, and fetal Thymus- associated lymphoid aggregates (as defined previously16). The evaluations were done with expert humans. A full example of the Visium processing pipeline and annotation is provided on the GitHub repository for this Article (see Code availability).
To process the Visium histology image data in higher resolution than the SpaceRanger defaults, we built a custom pipeline to get an additional layer of image resolution up to 5,000dpi, which is more suited for morphological analysis. We also developed our own fiducial image registration pipeline for increased accuracy where the fiducials are detected with cellpose v.2.1.1 and RANSAC from scikit-image v.0.22.0 is used for affine registration of reference fiducial frame (information provided by 10x Genomics). Lastly, for flexible tissue detection, we used Otsu thresholding with an adjustable threshold.
Source: A spatial human thymus cell atlas mapped to a continuous tissue axis
Mapping the thymus with STEMNET: high score progenitor cells for priming of mcTEC into mTEC(-Prolif)
The axis was placed in a sequential space as a reference for the entire thymus, using ten levels as the basis of the reference.
The STEMNET package50 (v.0.1) was used to determine the differentiation potential of mcTEC progenitor cells. The data was only retained for the purpose of this. The likelihood of each cell to adopt any of the possible fates was calculated with the purpose of setting maturation endpoints. The difference in the probabilities for the two fates for each cell was used to derive a high score for the priming of mcTEC(-Prolif) towards mTECI. The cells with a 0.5 or >0.50 score were labeled as mTECIprimed and the cells with a 0 or less score were labeled as unprimed.
Human fetal and paediatric data from several previous studies3,7,15,16 were included and reanalysed from raw fastq files. Details on sample processing, ethics and funding are available in the respective publications; details on the origin of each sample are provided in Fig. 1c and Supplementary Tables 1 and 2.
Metadata about scRNA-seq and CITE-seq samples, including information on source study, cell enrichment and donor age, are provided in Supplementary Table 1. Information about spatial data, including Visium, IBEX, RareCyte and RNAscope, is provided in Supplementary Table 2. The human genomes were mapped to with the help of the CITEsAQ data from the university. The human genome was mapped using the scRNA-seq data and 10x Visium data. All IBEX imaging was performed at the National Institute of Allergy and Infectious Diseases (NIAID), NIH. The non-ibEX datasets were generated at the same time as the IBEX. No fetal work was performed at the NIH and at Ghent University.
We used a network propagation approach to identify disease-relevant and cell cluster-specific subunits of our transcription factor network. differential expression-based marker gene scores for each cell cluster were propagated in same way, resulting in cell cluster specificity scores for each network node. For the multimodal analysis, we subsetted the full scRNA-seq dataset to include only paediatric data and removed T lineage cells without CITE-seq protein information.