Tumour evolution and microenvironment interact in 2D and 3D space
Getting Started with Xenium Panel Design: An Evaluation of Modulation and RNA Ligand Probe Selection Using the Seurat 68 Matrix
The module scores on top of each heatmap were calculated using a function from Seurat68. This score represents the average expression levels of a gene set. The score was calculated for each spot and a box plot was used to show the distribution of module scores in each microregion.
Custom Xenium gene and mutation probes were designed using Xenium Panel Designer (https://cloud.10xgenomics.com/xenium-panel-designer) following instructions outlined in the ‘Getting Started with Xenium Panel Design’ instructions (https://www.10xgenomics.com/support/in-situ-gene-expression/documentation/steps/panel-design/xenium-panel-getting-started#design-tool). In brief, 21-bp sequences flanking the targeted transcribed variant site were curated from the Ensembl canonical transcript (Ensembl v.100). All four possible ligation junctions (two for the WT allele and two for the variant allele, three in the case of deletions—two for the WT allele and one for the variant allele) were then evaluated. The sites where only non-preferred junctions were present were excluded. The two bases of the ligation junction sequence were the last base of the RBD5 (RNA binding domain) and the first base of the RBD3 probe. Preferred junctions were always prioritized over neutral junctions unless a neutral junction was necessary to avoid hairpins, homopolymer regions, dimers or an unfavourable annealing temperature. The temperature between 50 C and 70 C was set for the probes after adjusting their start lengths. Variant sites with probes that were predicted to form dimers or hairpins were excluded. Variant sites with homopolymer regions of five consecutive bases or more in either the RBD5 or RBD3 probes were excluded.
Identifying and Labeling Tumour Types Using Cell Segmentation and Alignment: A Computational Study for a SME tumour piece
We then use a gating procedure to identify cell types. To help fight differences between tissue types and images, thresholds are manually set for all the tissues used during cell typing. Above the intensity threshold, a positive representation was given for a certain marker, and a negative one. We then used the cell segmentation boundaries from the previous step to calculate the fraction of positive pixels for all cell typing markers in each cell. The feature matrix is a representation of the positive marker fractions for every cell in the sample. A cell was considered positive for a marker if >5% of its pixels were positive for that marker. Each sample was then labelled using a gating strategy. During gating, each cell was subjected to a series of AND gates, whereby if a cell passed all criteria for a given step, it was annotated as the cell type specified for that step, whereas if it failed the criteria it was passed on to the next downstream step in the gating strategy. The gating strategies used for the samples in this paper are presented in Supplementary Table 4.
The neighbourhoods classified as tumour neighbourhoods using the above criteria were considered to be positive voxels, and the other voxels were negative. The mask was smoothed with a 1.0 sigma kelvin. The input values were used to create a surface mesh for the tumour volume. We used the scikit-image implementation (skimage.measure.marching_cubes) of the marching cubes algorithm with default parameters.
The updated alignment tool PASTE70 was applied to enable partial image alignment. The tumour piece had its serial sections aligned with default settings. Each Visium data point in every ST section received new coordinates, denoted as x′ and y′, based on the alignment results. We then identified the nearest spot on each adjacent section for every spot, connecting them along the z axis. This process was used to link spots across the entire Z axis. We counted the connected spots after removing stromal spots, to see if one microregion was connected to another in an adjacent section. If any microregion on one section connected to the next section with more than three shared spots, then we considered these two microregions, located on different sections, as connected in 3D space and forming the same tumour volume. The connection was labelled as volume 1 and volume 2 in the figures. 9a–d.
The set of annotations were a combination of all possible cell types. For some images, not all proteins required to gate a specific cell type were present. For example, CD4 was not in every image panel and available to use in the annotation of CD4 T cells. The gating strategy was constructed to allow for the labels of cells as more general types if there is no specific T cell or CD4 T cell. If a cell was negative for all steps in the gating strategy, then it was labeled as unlabelled. The code for image format conversion and cell segmentation can be found at GitHub (https://github.com/estorrs/multiplex-imaging-pipeline).
Afterwards, we ran the Morph toolset (https://github.com/ding-lab/morph), which uses mathematical morphology to refine the tumour microregions. That is, if the total number of spots in a microregion is less than or equal to three, then we labelled all such spots as stroma. Last, Morph assigned the layer (for example, T1) of each spot of a tumour microregion by a sequence of mathematical morphology operations described in the Spot-depth correlation analysis method, which denotes the depth of a given spot inside a microregion.
Source: Tumour evolution and microenvironment interactions in 2D and 3D space
Graph-based clustering of integrated 4D neighbourhood volumes for HT397B1 and HT268B1 using CODEX, Slides and Slides
We had to sort out neighbourhoods with >50% overlap with copy number annotated subclones. We excluded the neighbourhoods that had less than ten total spots from the sample.
The Visium st spots were assigned to neighbourhoods in this manner. Each spot was assigned the neighbourhood label of the neighbourhood overlapping its spot centroid.
We integrated neighbourhood volumes for HT397B1 using a graph-based clustering approach. The neighbourhood voxel annotations were identified. A graph was then constructed, whereby nodes represented each neighbourhood partition combination, and edges are the distance (in the expression profile) between these partition combinations. The graph was then clustered to look for integrated neighbourhoods. The above clustering process has parameters provided in Supplementary Table 4. 3D neighbourhoods were displayed using an open-source visualization tool.
When neighbourhoods were assigned, the slides were used to create a 3D neighbourhood volume. For this, we used linear interpolation of neighbourhood assignment probabilities with the torchio library74.
After this, a slide token was concatenated to patch tokens. The slide token (representing the slide from which the image tile was selected) was indexed from a trainable embedding of size n_slides × d, where n_slides is the number of slides in the serial section experiment. The motivation for the slide token is that as it is passed through the transformer blocks, along with the patch tokens, information can be shared across all tokens, allowing the slide token to learn to attend to useful representations of the patches. The model was more robust due to this feature. The addition of the slide token made its way through the transformer blocks to the viT Encoder. All variables above and details of the transformer architecture are available in Supplementary Table 4.
Two separate runs of the model were trained for HT397B1 (six H&E, four CODEX and two Visium ST slides) and HT268B1 (four Visium ST slides). Training hyperparameters, such as number of training steps and batches, can be found in Supplementary Table 4. For HT268B1, only one instance was trained because only one data type was present. For HT397B1, three model instances were trained (one for each data type) and were subsequently merged following the procedure described in the section ‘3D neighbourhood construction and integration’.
Where λNBHD (maximum of 0.01) and λMSE (set to 1.0) are scalers for the neighbourhood loss (LNBHD) and reconstruction loss (LMSE), respectively. NBHD was raised from 0 to its maximum value during training.
lambda _rm NrmBrmHrmD
Source: Tumour evolution and microenvironment interactions in 2D and 3D space
Asymmetric vision transformers for enhancing the representation of image expression patterns in a hybrid image-to-image autoencoder model with alignment between adjacent sections
The autoencoder was trying to get better at two main tasks, the reconstruction of the expression profile of each image patch and the alignment of neighbourhood labels between adjacent sections. These two competing objectives forced the model to learn representative expression patterns while also keeping neighbourhoods aligned between input sections, which helped to combat neighbourhood differences due to batch effects. Differences in patch expression are quantified by the MSE and neighbours adjacency is enforced by not allowing cross-entropy of patches near each other.
The mean squared error (MSE) is one of the two main contributions to the overall loss function.
The neighbourhood annotation model consisted of an autoencoder with a vision transformer (ViT) backbone (Supplementary Fig. 7). An autoencoder is a training method for learning how input data are generated. Specifically, the network derives an approximation, Q, to the true posterior generating function, P, for the output, given the input. The autoencoder used was asymmetric, meaning that it wasn’t inverse copies of one another. The architectures described in theSupplementary Table four were similar to the one that was used for the encoder.
ViTs work on image tokens as input. In brief, image tokens are n-dimensional representations of patches of the input image. A uniform distribution of tile were collected from the input sections during training. 7a). The number of patches was determined by two parameters: patch height and patch width. Each patch was flattened to a 1, with the number of channels in the image being the number of genes. The patch was put into a matrix where the number of patches was taken from the tile. There is a token that represents the patch in the image tile. The linear layer projected the token shape to the dimensions of the transformer blocks.
The expression profiles for each patch were created in different ways. Expressions for CODEX and H&E patches were calculated as the average pixel intensities for each image channel over all pixels within the patch bounds. The expression profile of each spot within a Visium patch was linearly weighted by its distance to the centre of the patch. This differential weight helped to account for variation expression due to the number of spots that fall within patch boundaries.
We used Big Warp71, which came in the form of a software application. The first serial section is the fixed image and the second serial section is the moving image. After the second image was warped to the first image, the second image was used as the fixed image for the transformation of the third image. All images in the experiment were subjected to key point registration. A total of 4–20 key points were selected per image transformation. There was a moving field exported for each image transformation after key points were selected. This dense displacement field was then upscaled by a factor of 5 so it could be used to warp the full-resolution imaging data. The dense displacement field was used to register Visium data. There is a code used for registration that is available at this website.
The following changes took place before registration. Multiplex images were converted to greyscale images of DAPI intensity. The image was reduced by a factor of 5 before the key point selection. H&E images (also downsampled by a factor of 5) were used for keypoint selection with Visium data.
The original publication used a distance threshold of 1000 m to evaluate the spatial- based cell–cell interaction in the ST sample. The median sender and receiver signals for each interaction family were compared between all tumour boundary spots (including tumour boundary layer and TME boundary layer) and all non-boundary spots (Wilcoxon rank-sum test) on a sample. Boundary-enriched pathways with signal difference greater than 0.1 and FDR less than 0.05 are considered to be interaction pathways. Boundary DEGs were identified with FindMarkers function on three sets of comparisons: boundary/tumour, boundary/TME and boundary/all non-boundary. In the boundary test, a boundary DEG has adjusted the P value 0.25.
In the end, in the OCT workflows. 1a) , CalicoST simultaneously identified CNVs and groups microregions into spatial subclones. In the FFPE workflow, confident spatial CNV events in each microregion were first selected by comparing them with matching WES. A pairwise CNV similarity score was calculated across all the tumours. After using the function hclust, the micro regions were divided into clusters based on their similarity scores with the function cutree. Subclone assignments were manually reviewed to avoid overclustering and remove small outliers.
While non-negative matrix factorization can be used to identify cancer genes, it is a linear method that requires a constant and complete decomposition of the entire database, and it can be sensitive to variation. By contrast, Hotspot is based on gene–gene covariance, which better represents sets of genes that work together towards common functions, and is robust to batch effects57 (and is therefore likely to be more robust to variation between tumours). Importantly, Hotspot gene modules are based on covariance that can be localized to a cell subpopulation or exhibit potentially nonlinear graded expression over regions of the manifold. In cancer in particular, heterogeneous cell states adapted to different patient tumour contexts, such as metastatic sites, are likely to correspond to differences in gene covariance. Hotspot is also well suited to handle both gene pleiotropy and rare populations, which have important roles in tumour contexts. We therefore chose Hotspot to characterize sources of inter- and intra-tumour phenotypic heterogeneity in our dataset alongside global, manifold-defining modules of genes.
We focused on ten cases (comprising four BRCA, two CRC and four PDAC samples) with multiple spatial subclones for this analysis. To obtain subclone-specific DEGs, we used FindMarkers from the function in Seurat with the ‘wilcox’ test option DEGs between each subclone and TME. We then applied the cut-off for adjusted P < 0.01, average log2(fold change) > 1 and per cent expression in at least one cell type > 0.4 to select significant DEGs. To infer treatment response, we used the perturbation database LINCS L1000 (ref. 65), specifically the LINCS_L1000_Chem_Pert_down dataset from Enrichr66, to evaluate the gene set overlap between upregulated DEGs in spatial subclones and downregulated genes after compound treatment. The plot in supplementary figs. 4 and 5 was made after we sorted the data by odd.Ratio and picked top compounds from each subclone. The heatmap was added with the compound Metadata, including mechanism of action, which was obtained from CMap.io.
The DEG was designated as tumours-specific if it met both of the following criteria: first, it was a DEG when compared with all non-tumour cell types from at least one tumour subcluster, and second, it was the expression of the DEG.
The cell-type assignment was done based on the markers RAB32 and C. The breast’s normal epithelial cells were annotated with markers. Normal epithelial cells in the liver were annotated with the following markers: hepatocyte, ALB, CYP3A7, HMGCS1, ACSS2 and AKR1C1; cholangiocyte, SOX9, CFTR and PKD2. Normal epithelial cells in the pancreas, including ductal, acinar, islet-α, islet-β and islet-γ cells, were annotated with singleR (v.1.8.1) using reference data BaronPancreasData(‘human’).
The size of microregions and equilateral triangles for a WES-based CNV. A,E: A left versus right formula
We used the formula: density per m2 – n microregion per section – then divided by 8,660 m per spot. The density per m2 106 is determined by the number of microregions per millimeter.
The area was calculated using the spot size (55 m) and centre-to-centre distance between each spot (100 m). The Visium spots create a hexagonal lattice that covers the sample. The repeating unit of this lattice is a trapezoid shape centred at each spot’s centre that is composed of eight equilateral triangles. Half of the spot is the centre-to-centre distance for each triangle. Using the area equation of equilateral triangles and multiplying it by 8, we obtained the area of each trapezoid as 8,660 µm2, which is the average area occupied by each spot. To calculate the microregion size, we multiplied the spot count by 8,660 and divided by 106 to obtain the size in mm2.
When w_a is used, it means the window size from a WES-based CNV.
A,E, is a left versus right formula.
Source: Tumour evolution and microenvironment interactions in 2D and 3D space
Callmatic CNVs with GATK – I. Dataset analysis and modelling using the Bedtools 10X_S spatial dataset
GATK was used to callmatic CNVs. The hg38 human reference genome has been binned into target intervals using the PreprocessIntervals function and the interval-merging-rule of overLAPPING_ONLY. The panel of normals was created using the GATK functions CollectReadCounts and the minimum-median argument. The GATK function was used to count reads that overlap the target interval. Tumour read counts were denoised and standardized with the help of the GATK function. According to the GATK best practices, allelic counts for tumours were generated using the GATK function CollectAllelicCount. Segments were then modelled using the GATK function ModelSegments, with the denoised copy ratio and tumour allelic counts used as inputs. GATK uses a function called CallCopyRatioSegments to call copy ratios for segments.
The Space Ranger command was used to get the barcode matrix for each sample, with the help of the de multiplexed FASTQ files. Seurat was used for all subsequent analyses. The Load10X_Spatial function was used to create a Seurat object. The slide was normalized and scaled to correct for effects. The same scaling and normalization methods were used for every merged analysis or subsequent subsetting of cells. Spots were clustered using the original Louvain algorithm, and the top 30 principal component analysis dimensions using the FindNeighbors and FindClusters functions as described in the ‘Analysis, visualization, and integration of spatial datasets with Seurat’ vignette from Seurat (https://satijalab.org/seurat/articles/spatial_vignette.html).
Bedtools62 intersection was used to map copy number ratios from segments to genes and to assign the called amplifications or deletions. The weighted copy number ratio of each overlap segment was calculated by using the CallCopyRatioSegments function in a custom Python script. If the resulting z score cut-off value was within the range of the default z score thresholds used by CallCopyRatioSegments (v.0.9,1.1), then the bounds of the default z score threshold were used instead (replicating the logic of the CallCopyRatioSegments function).
Somatic mutations were called from WES data using the Somaticwrapper pipeline (v.2.2; https://github.com/ding-lab/somaticwrapper), which includes four different callers: Strelka (v.2.9.10)54, MUTECT (v.1.1.7)55, VarScan (v.2.3.8)56 and Pindel (v.0.2.5)57. We kept exonic single nucleotide variant (SNV) called by any two callers among MUTECT, Var Scan and Strelka. The minimal coverage cut off was applied for the merged SNVs and indels. We also filtered SNVs and indels by a minimal VAF of 0.05 in tumours and a maximal VAF of 0.02 in normal samples. We also filtered any SNV within 10 bp of an indel found in the same tumour sample. The rare mutations were found by using an established gene consensus list. In a previous study, we reported that we use COCOON to combine adjacent SNVs into double-nucleotide polymorphisms using Somaticwrapper.
Images of OCT-Embedded and FFPE Tissues using the Visium Spatial Protocols-Tissue Preparation Method
Tumour fragments were transferred into a tube which had a pre-filled 5ml of IGFF medium supplemented with antibiotics and a commercial cocktail of tissue digestion agents (tumour Dissoci). Tumours were eaten using the gentleMACS Octo Dissociator, which can be used for 30 minutes. Considering the heterogeneity of tissue specimens with respect to cell viability, immune infiltration, blood content, necrosis and calcification, the digestion state of the tumour fragments was assessed every 10 min under an inverted microscope. Digestion was interrupted before 30 min if at least 50% of the tumour material appeared broken into clusters of 1 to 10 cells. Next, cell cluster solutions were filtered through a 100 μm cell strainer, and washed three times with DPBS supplemented with antibiotics, with each centrifugation step performed at 100g for 3 min at room temperature. The final cell suspension was washed and removed from the environment for 5 min at 500g and 4 C.
OCT-embedded tissue or FFPE tissue samples were sectioned and placed on a Visium Spatial Gene Expression Slide following the Visium Spatial Protocols–Tissue Preparation guide. The samples that were part of the serial section were collected with an interval from 5 to 100 m. When doing serial sectioning, the first section was named as U1, followed by U2, U3, and so on. Selected sections were loaded onto Visium slides and the distance between each section was recorded. Detailed methods have been described in a previous publication. In brief, fresh tissue samples were coated with room temperature OCT without any bubbles. Blocks were scored into a suitable size for capture areas and then they were sectioned into smaller sections after the H&E staining was applied to the tissue samples. Sections were then fixed in methanol, stained with H&E and imaged at ×20 magnification using the bright-field imaging setting on a Leica DMi8 microscope. Following the user guide for Visium Spatial Gene Expression Reagent kits, ST libraries were constructed using tissue samples. cDNA was reverse transcribed from the poly-adenylated messenger RNA, which was captured using primers on the slides. The second strand was denatured from the first strand. Free cDNA was then transferred from slides to tubes for further amplification and library construction. The libraries were on the S4 flow cell. For FFPE samples, detailed methods have been described in a previous publication47. Quality control was done by evaluating the quality of the DV200 ofRNA that was taken from the FFPE tissue sections and then performing the Tissue Adhesion Test described in the 10x Genomics protocol. Sections 5 m were placed on the Visium Spatial Gene Expression Slide. After overnight drying, slides were incubated at 60 °C for 2 h. The Visium Spatial protocol was used for the deparaffinization. Sections were stained with H&E and imaged at ×20 magnification using the bright-field imaging setting on a Leica DMi8 microscope. Afterwards, decrosslinking was performed immediately for H&E stained sections. The human whole transcriptome probe panels were put in the tissue. After these probe pairs hybridized to their target genes and ligated to one another, the ligation products were released following RNase treatment and permeabilization. The ligated probes were then hybridized to the spatially barcoded oligonucleotides on the capture area. ST libraries were generated from the probes and sequenced on a S4 flow cell of an Illumina NovaSeq 6000 system. Relevant protocols can be found at protocols.io (https://doi.org/10.17504/protocols.io.x54v9d3opg3e/v1 and https://doi.org/10.17504/protocols.io.kxygx95ezg8j/v1)48,49.
To maximize capture of all areas of the tumour including the invasion front, full clinical pathology sections prepared for clinical diagnosis were used for imaging (not core punches as typically used for tissue microarrays). Primary antibody staining conditions were optimized using standard immunohistochemical staining on the Leica Bond RX automated research stainer with DAB detection (Leica Bond Polymer Refine Detection, DS9800). Using 4 µm FFPE tissue sections and serial antibody titrations, the optimal antibody concentration was determined followed by transition to a seven-colour multiplex assay with equivalency. Optimal primary antibody stripping conditions between rounds in the seven-colour assay were performed following one cycle of tyramide deposition followed by heat-induced stripping (see below) and subsequent chromogenic development (Leica Bond Polymer Regine Detection, DS9800) with visual inspection for chromogenic product with a light microscope (T.J.H.). Supplementary Table 8 describes multiplex assays.
CellBender: a method of removing ambient RNA from genomic data using a probabilistic model for cell-by-gene analysis
CellBender is a method of removing ambient RNA from data. It starts with levels of ambient RNA and rates of barcode swapping per gene and droplet, from a cell-by-gene count matrix. This probabilistic model is then used to generate a denoised (that is, ambient RNA-corrected) count matrix, as well as the probability that each droplet contains a cell, which can be used for calling real cells. To run CellBender we used the following parameters: (1) set the number of cells as the number of cells loaded into each 10x Chromium lane per sample, typically 5000– We used the denoised count matrix from CellBender for all of our analyses.
About 100–250 ng of genomic DNA was fragmented on a Covaris LE220 instrument targeting 250-bp inserts. Automated dual-indexed libraries were constructed using a KAPA Hyper library prep kit (Roche) on a SciClone NGS platform (Perkin Elmer). Up to ten libraries were pooled at an equimolar ratio by mass before the capture. The library pools were hybridized using xGen Exome Research Panel v.1.0 reagent (IDT Technologies), which spans a 39-Mb target region (19,396 genes) of the human genome. The libraries were hybridized for 16-18 h before being washed to remove spuriously hybridized library fragments. Enhancing library fragments was done to prevent overamplification. The enriched libraries were amplified using KAPA HiFi master mix (Roche) before sequencing. The concentration of each captured library pool was determined through qPCR using a KAPA library Quantification kit according to the manufacturer’s protocol (Roche) to produce cluster counts appropriate for the Illumina NovaSeq-6000 instrument. Next, 2 × 150 paired-end reads were generated targeting 12 Gb of sequence to achieve around 100× coverage per library.
Source: Tumour evolution and microenvironment interactions in 2D and 3D space
Alignment of FASTQ Files with BWA-MEM for Whole-Genome Detection and Allele-Specific Copy-Number Anomalies
All data analyses were conducted in R and Python environments. Details of specific functions and libraries are provided in the relevant methods sections above. Significance can be determined using the Wilcoxon rank-sum test, proportion test or Pearson correlation test. Significant is the P values 0.05. There are details of statistical tests in the figure legends.
The files were preprocessed using trimGalore and had parameters set to default. FASTQ files were then aligned to the GDC’s GRCh38 human reference genome (GRCh38.d1.vd1) using BWA-mem (v.0.7.17) with parameter -M and all others set to default. The output SAM file was converted to a BAM file with parameters -Shb and all others set to default. BAM files were sorted and duplicates were marked using Picard (v.2.6.26) SortSam tool with the following parameters: CREATE_INDEX=true, SORT_ORDER=coordinate, VALIDATION_STRINGENCY=STRICT, and all others set to default; and MarkDuplicates with parameter REMOVE_DUPLICATES=true, and all others set to default. All parameters had been set to default for the index that would be used to index the final BAM files.
The FACETS allows for allele-specific estimates of copy-number alterations at both the genes and the chromosomes. FaceTS was used to detect whole-genome duplication events, to infer the clonality of somatic mutations and to assess arm level copy-number changes.
Four samples were technically duplicated in order to determine the error rate. The minimum VAF threshold for downstream filtering was chosen so that duplication of mutation calls in technical replicates was achieved. It was found to be 0.02. A nonsense mutation with 100 read depth was the only one which was excluded from the calls.
FASTQ files were aligned against the Genome Reference Consortium mouse genome 39 (GRCm39)59 using BWA-MEM (https://github.com/lh3/bwa). VarDict was variant caller and there was a minimum allel fraction threshold of 0.01 for the call. There was a variant annotation done. The list of called variations had to be removed because they weren’t pass internal noise filters. The removal of indels happened because of the ENU’s tendency to cause single-nucleotide variant 61. Finally, variants were retained only if they were called in at least two amplicons per sample and supported by at least five mutant reads. Given that codons 73–84 and 122–139 of Apc were only covered by one amplicon, variants falling in these regions were manually inspected, and only retained if they were called by at least five mutant reads and a VAF of more than 0.01. Only one sample was affected by this.
NEB Q5 High-Fidelity DNA polymerase (M0491S), with primers hybridizing to exons 4 and 16 of Apc (for_261-AAAAATGTCCCTTCGCTCCT and rev_3149-CTGTGAGGGACTCTGCCTTC, respectively) was used for PCR amplification of cDNA template. For each sample a different five nucleotide barcode was incorporated onto the 5′ end of the forward primer. The size distribution of the sample is confirmed by analyzing the gel electrophoresis products of the sample and then pooled into an equimolar ratio. The PacBio SMRTbell library was built and tested on a cell from the Earlham Institute.
There were micro-dissected minor tumours that were isolated with the help of the mRNA. According to the instructions of the manufacturer, the first strand of cDNA synthesis was done using a NEB Protoscript II kit. It was possible to use both oligo-dT or a gene specific reverse primer.
The iScript kit was used to make the cDNA from 1 gRNA. The manufacturer’s instructions on aQuantStudio 6 are what provided the instructions for the real-time quantitative PCR for Notum. Relative fold change in gene expression was calculated using the ({2}^{-{{\rm{\Delta \Delta }}C}_{{\rm{t}}}}) method. All ΔΔCt values were normalized to the housekeeping genes Gapdh (Mm99999915_g1) and Rpl37 (Mm00782745_s1).
Skoufou-Papoutsaki et al. 73 described how a knockout of Apc was performed. In brief, mouse small intestinal organoids were single-cell dissociated at 37 °C in TrypLE Express (Gibco, 12605010) and Y-27632 (1:1,000, Tocris, 1254) for 30 min. One-hundred thousand cells were then put into a vat of Invitrogen and single guideRNA complex. sgRNAs were designed using Benchling (https://www.benchling.com/) and Indelphi (https://indelphi.giffordlab.mit.edu/)74. Guides were intended to lead to the out of frame indels at codons S 96 and T619. The sgRNA sequences are pre-Armadillo and Armadillo. The cells were then transferred into a 16-well nucleovette strip (Lonza) and incubated for 10 min at room temperature. Electroporation was performed on an Amaxa 4D Nucleofector (Lonza) using the DS138 programme. After 10 min at 37 °C, cells were transferred to a 0.5 ml tube, suspended in 20 μl Cultrex and plated as described above. Once organoids were formed (usually about 7–10 days after plating), single organoids were picked using a EZGrip micropipette (CooperSurgical Fertility Solutions) under microscopic visualization. Single-picked organoids were then placed in 5 μl TrypLE Express for 10 min at room temperature, mixed with 15 μl Cultrex and replated to generate clonal organoids. According to the instructions provided by the manufacturer, PicoPure DNA had been used to successfully extract genetic material. The custom designed primers and product were used. PCR primers used were: Pre_Armadillo forward, GGCAGATGGGTTCAAAGGGGTAGAG; Pre_Armadillo reverse, AAACTCCCACGCACACACAGTACTT; Arm forward, TGACTCATAGAAACAGCACTGACCCA; Arm reverse, GCATGGCTGGATTTCTCAACTACCA; MCR forward, TCAGACAACACAGGAAGCAGA; and MCR reverse, GGCCCACTCTCTCTCTTCTC. The knockout score and clonality were determined using the ICE Synthego platform.
Micro-dissected mouse intestinal tumours were immediately placed in 200 μl dissociation medium at the time of dissection. They were put into 37 C for 30 minutes. Next, 5 ml ice-cold ADF was added and the sample was spun at 1,200 rpm at 4 °C and supernatant aspirated. The pellet was resuspended in 5 ml The ADF spun down again. The pellet was placed in a small volume of the cultrex basement extract. This was plated and left to set at 37 °C for 10 min before the addition of growth media. Organoids were grown in warm weather at a temperature of 37 C.
Standard growth medium consisted of advanced DMEM/F12 (ADF) (Invitrogen, 12634-028) with 10 mM penicillin-streptomycin (Gibco, 15140122), 10 mM l-glutamine, 10 mM HEPES, N-2supplement, and B27 supplement are included in the list. Dissociation medium was made with DMEM (Gibco, 11965092), 2.5% FBS, 10 mM The penicillin-strepomycin, 50 g ml1 liberase, and 0.8 g ml1 were all listed by Worthington Biochem. The Stem Cell Tech has a DNAse I.
Bioinformatics assessment of RNA-sequence interrogation datasets for gene expression quantification and RNA subtyping
Sequence read quality was assessed using FastQC (v0.11.9; http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Adapter content was trimmed from the reads using Trimmomatic (v0.39)62. Trimmed reads were aligned to a certain point. Quality control of aligned reads and Ensembl release 103 for quality control were carried out using Picard tools. Salmon was used to carry out Gene expression quantification against Gencode Mouse release M30 The DESeq2 package64 was used for differential gene expression. The adjusted P value was used to determine how genes were differentially expressed. The Gene set enrichment analysis was performed using the GSEA function. Signature scores were based on the Hallmark Pathways66 and published gene sets for mouse ISCs by Muñoz et al.67, Merloz-Suarez et al.68 and mouse small intestinal and colonic secretory signatures from Tomic et al.69. The Wnt pathway, APC knockout genes, and a list of genes for the mouse in the intestinal region were used. The signatures were derived from a compendium of single-cell RNA-sequencing experiments. The MmCMS package 24 contains genes set collection C, which is used for consensus genetic subtyping. The default prediction probability of 0.6 was used for the pathway-derived subtyping.
The target library was prepared using the Standard BioTools protocol and the 8.8.6 integrated fluidic chip. In brief, each IFC allowed the highly multiplexed interrogation of 48 samples against 8 independent panels of primers, leading to the generation of 286 amplicons for each sample. The amplicons from the IFCs were quantified using a Bioanalyzer. Sequencing was performed as paired end 150-bp reads on the Illumina platform.
Standard BioTools’ D3 Assay Design software was used to design a targeted panel of primers covering ten genes (Apc, Ctnnb1, Kras, Nras, Hras, Braf, Pten, Fbxw7, Smad4 and Trp53). The coverage for all of the genes was limited to previously identified hotspots on an exome hybridization panel. All the targeted nucleotides in the panel were covered by at least two amplicons apart from Apc and Pten, which had 99.2% and 77% dual coverage, respectively.
DNA extraction from bulk or micro-dissected tumours was performed using a QIAmp DNA FFPE Tissue kit (Qiagen, 56404) according to the manufacturer’s instructions, apart from a longer lysis incubation time of 12 h at 56 °C and omission of the 90 °C incubation step. The results of the analysis were used by aNanodrop spectrophotometer. Extracted DNA was stored at −20 °C.
Mice were culled by cervical dislocation. The whole intestine was dissected, flushed with cold PBS, cut longitudinally, and wholemounted. After fixation in 4% paraformaldehyde for 24 h at 4 C the tissue was washed in PBS and randomly chosen portions of the bowel were excised. CuBIC protocol 56 was used for optical clearing. In brief, excised segments were incubated with CUBIC-1a solution (10% urea, 5% N,N,N′,N′-tetrakis(2-hydroxypropyl) ethyl-enediamine, 10% Triton X-100 and 25 mM NaCl in distilled water) at 37 °C for 7–10 days with alternate day solution changes. It was used for counterstaining at a ratio of 1:1,000. The cleared tissue was then washed in PBS for 24 h. Rapiclear 1.52 was used for additional clearing and Refractive index matching. Finally, the samples were mounted in a 0.25 mm i-Spacer (Sunjin Labs) for confocal imaging.
All histological quantification was performed using QuPath (v.0.4.3; https://github.com/qupath/qupath)58. The first manually createdAnnotations based on Confetti status were used to create the first Heterotypic tumours. Positive cells for other markers were identified using the positive cell detection feature with an intensity threshold of 5 and a nucleus background radius of 8 m. Results were reported as the number of positive cells per unit area of the annotated area if the staining was chromogenic or fluorescent. Ki67 results were reported as percentage of DAPI-positive cells.
ACD used the 2.5 LS Duplex Reagent Kit to perform simultaneous detection of Lgr5 and Anxa1 along with Notum. Three- Micrometre thick sections were baked at 60 C before being loaded on to the Bond RX instrument. Slides were deparaffinized and rehydrated on board before pre-treatments using Epitope Retrieval Solution 2 (AR9640, Leica Biosystems) at 95 °C for 15 min, and ACD Enzyme from the Duplex Reagent kit at 40 °C for 15 min. A signal amplification was done according to the manufacturer’s instructions. The Bond Rx was used to perform the red detection of C2 according to the ACD protocol. Slides were then removed from the Bond Rx and detection of the C1 signal was performed using the RNAscope 2.5 LS Green Accessory Pack (ACD, 322550) according to kit instructions. VectaMount Permanent mounting Medium was mounted on the slides, dipped in Xylene and heated to 60 C. The images on the slides were created using the software on the aperio AT2. Images were captured at 40× magnification, with a resolution of 0.25 μm per pixel.
Images were acquired on a Leica SP5 TCS confocal microscope (LAS software v2.8.0, Leica) with a 10× objective, 1.4–1.7 optical zoom and 8–12 μm z-steps throughout the whole thickness of the tissue. The image analysis was done using a software. All identified tumours had their Confetti status manually assessed at all of the acquired z positions. If a tumours had at least two Confetti colours or one Confetti colour, and no unlabelled glands, it was deemed to be Heterotypic. They most probably represent entrapped normal crypts, so they were not used for the purpose of determining heterotypic status.
The cause of the start of the oncogene fields was caused by a single injection of 4% (Merck T5668) dissolved in syrup and/or oil. Chemical mutagenesis was performed exactly 10 days after field induction using 200 mg kg−1 ENU dissolved in ethanol/phosphate-citrate buffer (1:9) given intraperitoneally.
Male and female mice of at least 8 weeks of age were used for the experiments. The Federation of European Laboratory Animal Science Associations recommend that mice be housed in a pathogen-free facility, with individually ventilated cages, under controlled conditions, which include temperature, humidity, and light/dark cycle. They had food and water. The mice had not been involved in any procedures prior to the study. For survival curve generation, the mice were aged until they showed pre-defined clinical signs of tumour burden (anaemia, hunching, and loss of body condition). No mice were allowed to go over the endpoints. No randomization or blinding was used. The sample sizes were determined using the results of preliminary experiments. All animal experiments were performed in accordance with the guidelines of the UK Home Office under the authority of a Home Office project licence (PD5F099BE) approved by the Animal Welfare and Ethical Review Body at the CRUK Cambridge Institute, University of Cambridge.
Source: Polyclonality overcomes fitness barriers in Apc-driven tumorigenesis
Simulated growth of bowel tumours using the spatstat package and statistical analysis of C57BL/6 Mn micrographs and RNA sequences
The inducible Cre line was crossed on a C57BL/6 background. The LSL-KrasG12D54 was used in some experiments. Genotyping was performed by Transnetyx using real-time PCR.
A script was written to randomly assign a Confetti label to crypts in a 10 100 field of crypts. The number of crypts with the same fluorophore was quantified for each simulation, as well as the distribution of coloured crypts in patch sizes.
A simulation was made using the observed tumours growth rates in the Apchet + ENU model. A number of points were put into t plus 0 after ENU with each point attributed a Confetti label and growth rate from the distribution of frequencies. These points were then allowed to expand until the simulation was stopped at the humane endpoint reached by the mouse. The number of collisions resulted in tumours for every simulation. The simulations were repeated for 10,000 seeds for each segment of intestine scored. A comparison of the observed number of tumours to expected number was done using a two-tailed t-test.
To investigate whether heterotypic tumours arise in regions of highest density, the spatstat package was used to calculate local spatial tumour density within each imaged bowel segment. The local spatial density was extracted at the location of each tumour, and densities at heterotypic tumours were compared to those of non-heterotypic tumours using a Q–Q plot and the Kolmogorov–Smirnov test. If clustering occurs in high density regions, the density distributions would be higher on average at Heterotypic tumour locations, according to the hypothesis.
A mixed-effects model was built using the package nlme to quantify the growth dynamics of different types of tumors. The rate of growth was quantified during the growth phase, which is between 40 and 63 days after ENU.
Visualization and statistical analysis of data were performed in the R statistical computing environment (version 4.2.3) or GraphPad Prism version 10.2.2 (341). Multiple testing correction of P values was conducted using the Benjamini–Hochberg method. All immunostaining and RNAscope experiments were performed on at least three independent biological replicates (three different mice). Micrographs depict representative data derived from at least three independent biological replicates. The figure legends and figures contain statistical tests and corresponding P values. Box plots display the distribution of data using the following components: lower whisker show the smallest observation greater than or equal to lower hinge minus 1.5× IQR; lower hinge shows the 25% quantile; the centre line shows the median, 50% quantile; the upper hinge shows the 75% quantile; the upper whisker shows the largest observation less than or equal to upper hinge plus 1.5× IQR.
We applied MAGIC (v.3.0.0) imputation59 to normalized, log-transformed count matrices to denoise and recover missing transcript counts due to dropout. Imputation was performed using conservative parameters (t = 3, ka = 5, k = 15). The main text and figure legends are used to describe the values used for visualization of gene expression and gene signature expression.
The chart review identified patients who had signed pre-procedure consent to the IRB protocols and were selected for the study. There was no statistical method used to get a sample size. Freshly cut surgical tissue was processed into single-cell suspensions for analysis and generated organoids, if enough tissue was available. The portion was fixed in formalin after being embedded in paraffin. Tissue was generally processed within 1 h of surgical resection. Archival formalin-fixed, paraffin-embedded (FFPE) clinical tissue blocks for immunostaining were identified by database search and chart review. An expert gastrointestinal pathologist supervised the processing and interpretation of tissue data. Where the trio of normal colon, primaryCRC and metastaticCRC were collected, the patient was tracked longitudinally through their clinical course atMSK with the collection of tissue surplus to diagnostic requirements.
The manual review of patient electronic medical records by board certified medical oncologists were collected as part of institutional review board approved protocols. The time to each event was calculated from the date of diagnosis in order to compare across patients. The data was collected and managed using REDCap electronic data capture tools. 17 out of 31 patients had multiple metastatic sites at the time of surgery, and had >50% of tumour sites remaining after surgery. 17 out of 31 patients had early-onset CRC (age of diagnosis, <50 years). Clinical MSK-IMPACT targeted exon sequencing was performed on tumour/normal tissue from 27 out of 31 patients and revealed expected mutations53 (Extended Data Fig. 1c). Consistent with the low percentage (<5%) of metastatic CRC that is mismatch repair deficient/microsatellite instability high, only one patient in our cohort had an microsatellite instability indeterminate tumour. Clinical data collection was censored on 30 September 30 2022.
We collected 50–300 liters of freshly resected surgical tissue in 5 liters of the IGFF organoid medium. For primary and metastatic tumours, specimens were placed into a 15 cm Petri dish using sterile forceps and washed three times with DPBS (Thermo Fisher Scientific) supplemented with the above-described antibiotic cocktail, and minimally chopped with sharp sterile blades to enable transfer of tumour fragments using a pre-wet 25 ml serological pipette.
The tissue was put into a tube with dissociation buffer and then put into it’s own container. For a maximum of 30 minutes, mucoal fragments were put in a petri dish and gently turned at 4 C. Every 10 min, tissue fragments were evaluated under an inverted microscope. If at least 30% of the mucosal material appeared broken into clusters of 1 to 5 colonic crypts, dissociation would be stopped before 30 min. The crypt solution was first sieved through a 1 mm cell strainer to separate crypts from large chunks of undissociated mucosa. A volume of DPBS was supplemented with antibiotics. At this point, the 1 mm filter was flipped and inverted into a fresh 50 ml tube. Up to 25 ml of DPBS supplemented with antibiotics was flashed through the inverted filter to recover the undissociated mucosal tissue. After manually shaking the suspension of mucosal tissue fragments (approximately 5 times), the collection of clusters of colonic crypts was reattempted as described above. Based on the iteration of filtration and manual agitation steps, up to three additional fractions of crypt suspensions were collected. Each centrifugation step took 100g for 3 min at room temperature after the Crypt suspensions were washed three times. If the individual crypts were assessed to have low crypt content, the suspensions would either be processed separately or pooled together.
For both tumour and normal tissue, if blood traces were visible under an inverted microscope, the cell pellet was resuspended in 1–5 ml ACK lysis buffer (Lonza), according to the pellet size and incubated for 5 min at room temperature. Quenching was performed with three volumes of DPBS supplemented with antibiotics, followed by an additional wash to remove ACK traces. The pellet was further processed for either scRNA-seq or organoids. A high quality viable single-cell suspension can be used for downstream analyses.
For validation, organoids underwent targeted exome sequencing by MSK-IMPACT53 and key oncogenic genomic alterations were identified by OncoKB55 (see below). Diagnostic tissue from originating tumours was sequenced to confirm that these alterations were conserved in each derived organoid line. Organoids were verified on the basis of short tandem repeats at the time of establishment and before every experiment, and were routinely tested for mycoplasma contamination (MycoALERT PLUS detection kit, Lonza).
The patient samples were processed using the default parameters and platform of 10x Genomics. A preliminary count matrix of cells is produced when the SEQC performs read demultiplexing, alignment and Umi. By default, the pipeline will remove putative empty droplets and poor-quality cells based on (1) the total number of transcripts per cell (cell library size); (2) the average number of reads per molecule (cell coverage); (3) mitochondrial RNA content; and (4) the ratio of the number of unique genes to library size (cell library complexity). The sensitivity to dissociation of the colorectal epithelium allowed us to see increased indicators of cell stress, apoptosis, and dropletContamination in many samples, which can obscure the existence of meaningful biological gene expression. As such, typical ad hoc cell filtering based on identifying a steep dropoff in the number of transcripts per droplet (that is, a deviation leading to a ‘plateau’ in ambient RNA levels), could impair the extraction of meaningful biology. We therefore sought to systematically evaluate and correct for ambient RNA expression and filter for real single cells using CellBender (v.0.1.0)58 as described below.
The set of genesconstituting each module has to be consistent with the number of features, and we used minimum_gene_threshold set to 20 and core_only set to make it happen. We then calculated Pearson correlations between the module scores of each set of Hotspot modules and the set obtained with 2,000 HVGs, as used in this study. The best-matching module is the most correlated with the original module in the study. When you look at every set of gene features, there is a subset of modules that show close correspondence to our final set of modules.
Next, the nearest neighbour and between-case maps were converted to within-sample and between-case affinity matrices. The resulting matrices were concatenated to obtain an augmented cell–cell affinity matrix that consists of three main components: (1) similarity between in vivo cells; (2) similarity between in vitro cells; and (3) similarity between in vitro and in vivo cells. This matrix was used to input the classification for PhenoGraph to be used to display labels from the reference to the unlabelled dataset.
Synthesis and PCR amplification of de novo mirE shRNA sequences for doxycycline inducible PROX1 knockdown experiments
For doxycycline inducible PROX1 knockdown experiments, de novo 97-mer mirE shRNA sequences were synthesized (IDT Ultramers) and PCR amplified using the primers miRE-Xho-fw (5′-TGAACTCGAGAAGGTATATTGCTGTTGACAGTGAGCG-3′) and miRE-EcoOligo-rev (5′-TCTCGAATTCTAGCCCCTTGAAGTCCGAGGCAGTAGGC-3′) as described previously83. Sequences (shPROX1-1: TGCTGTTGACAGTGAGCGCGAGGACCAAGATGTCATCTCATAGTGAAGCCACAGATGTATGAGATGACATCTTGGTCCTCATGCCTACTGCCTCGGA; shPROX1-2: TGCTGTTGACAGTGAGCGCCCCCGAGAAAGTTACAGAGAATAGTGAAGCCACAGATGTATTCTCTGTAACTTTCTCGGGGATGCCTACTGCCTCGGA) were cloned into the LT3GEPIR backbone83 (Addgene, 111177) and used to generate lentiviral particles to transduce into organoids as described previously82. The control was made using the original plasmid containing an shRNA sequence. The organsoids were selected through the use of the medium supplemented with puromycin. Organoids were isolated and plated at a density of 2000 cells per 40 l of Matrigel and maintained in a mixture of HISC or IGFF, with 2 g of liquid drake added to each cell. For an experiment, organoids containing PROX1 and control shRNA were cultured in a medium supplemented with 2 gml1 for 7 days and stained with DAPI.
The Matrigel contained about 4,000 organoids that were recovered using 3 mM. The lysed EDTA was washed, and lysed with a 1 RIPA buffer supplemented with PPI and benzonase. The concentration of the kDa was determined using the Pierce BCA test. A total of 10 μg protein per sample was separated by SDS–PAGE on Bis-Tris polyacrylamide gels (Thermo Fisher Scientific, NW04120BOX), transferred to activated PVDF membranes (Millipore, IPFL00010) and blocked in 3% BSA-TBST solution for 30 min. The membranes were incubated overnight at 4 °C with the following antibodies: mouse anti-β-actin (1:1,000, Thermo Fisher Scientific, AM4302) and rabbit anti-PROX1 (1:1,000, Abcam, ab199359), followed by secondary antibody incubation with 488 anti-mouse and 680 anti-rabbit secondary antibodies (1:5,000, LI-COR Biosciences, 1 h, room temperature) before imaging (Odyssey CLx). Western blotting was quantified using ImageJ.
We used Mesmer (v.0.12)81, a deep-learning cell segmentation algorithm, to identify cell boundaries in all COMET and Vectra images. The input to Mesmer (v.0.12) is a single nucleus-stained image and a single membrane or cytoplasm-stained image to define the extent of each nucleus and cell. We used DAPI as a nuclear marker for all COMET and Vectra images. The channels for multiple cell types were combined into a single image by min-max scaling, using the MinMaxScaler function in the preprocessing. For COMET, we combined CK20, HER2, CK5, SYNC (normal and tumour epithelial cells) and VIM (stromal cells). HER2, SOX2, CK20, CDX2 and CHGA were used for Vectra panel 1. For Vectra panel 2, we used TP63, OLFM4, TROP2, and the like.
An experienced doctor annotated 167 FVs for high background signal to determine if they have a high signal. To identify high-background images in the remaining 435 FOVs, we first found the highest level of background CK5 signal (10th percentile of expression across all cells in the FOV) within FOVs labelled low-background (around 0.0068). The Fovs were classified as high background because of a background signal of >0.05)68. As a result, 327 unlabelled FOVs with low background were used in later analyses, and 108 with high background were removed. We used 456 FOVs with a low level of CK5 background expression in figures relating to the Vectra panel 2.
The levels of marker expression are determined by summing the brightness values over all the cells. To ensure that our downstream analysis was not influenced by cell size, we then divided the per-cell expression for each channel by the cell boundary sizes determined by the regionprops function, as described above. All cells were pooled together into cell-by-expression matrices within the same panel (Vectra panels 1 and 2 and COMET) for downstream analyses.
We use the region props function in the Python skimage package to estimate the cell size, eccentricity, and centoid of each cell boundary. We used subsampling to make the COMET images fit in the system’s memory. For both cell size and DAPI expression, we found the distributions of segmented cells produced by Mesmer (v.0.12) were bimodal and the lower mode contained primarily empty regions and not real cells. We therefore filtered out all predicted cell boundaries below threshold values of 30 px2 cell size (estimated from the distribution) and a log2-normalized DAPI intensity of 11 (COMET), 1 (Vectra panel 1) and 1 (Vectra panel 2) (estimated from the distribution). This resulted in a COMET dataset of 6,867,000 cells across 18 Fivs and a Vectra panel 1 dataset of more than 6 million cells across 661 Fivs.
We needed to summarize our classification using coarser cell typing since there are large differences between in vitro and in vivo samples. Thus, to summarize our organoid sample classifications, we aggregated the probabilities of different cell states into three generalized categories; ISC-like, combining TA/Proliferative for ISC/TA, absorptive-like and secretory-like for differentiated intestine, and combining fetal/injury repair, neuroendocrine and squamous for non-canonical. These groupings can be used to determine if a cell is from any of the cell states which were combined. The probabilities for each resulting categories are plotted using the python-ternary (v.1.0.8)79 package (Fig. 3b and Extended Data Fig. 10b).
We aimed to generate a ranked list of human transcription factors that are associated with the fetal progenitor state in a conserved manner across non-canonical patient tumours. Starting with a list of 1,665 human transcription factors80, we restricted potential targets to transcription factors for which we observe (1) greater than 10 UMIs total in all four patient datasets (leaving 1,099 transcription factors); (2) at least 5 UMIs in any cell in all four patient datasets (leaving 527 transcription factors); and (3) at least 50 transcription factor-expressing cells in all four patient datasets (leaving 508 transcription factors). The plan was to remove transcription factors that are not easy to target and to restrict our analysis to factors that are more reliable in their correlation with the fetal signature.
The log-transformed fold change between irinotecan and other conditions was used to figure out the treatment response for the remaining six transcription factors. log-transformed fold changes were calculated from single-cell data only using cells classified as non-canonical (see the ‘Mapping organoid data to patient tumour’ section) (Extended Data Fig. 11b).
Source: Progressive plasticity during colorectal cancer metastasis
Multispectral imaging of seven-colour multiple x-stained slides with the Vectra Imaging System: identification of seven marker channels using the PhenoGraph
Seven-colour multiplex-stained slides were imaged using the Vectra Multispectral Imaging System version 3 (Akoya). Scanning was performed at ×20 (×200 final magnification). Filter cubes used for multispectral imaging were DAPI, FITC, Cy3, Texas Red and Cy5. A spectral library containing the emitted spectral peaks of the fluorophores in this study was created using the Vectra image analysis software (Akoya). The multispectral images from the single-stained slides allowed for identification of seven marker channels of interest using Inform v. 2.4 image analysis software, as the library was separate from each multispectral cube into individual components.
The augmented affinity matrix was supplied from step 2 to the PhenoGraph. This function converts the affinity matrix into a row-normalized Markov matrix and uses it to calculate the probability of random walks from unlabelled cells to a class of labelled cells. The maximum probability is assigned to each unlabelled cell.
Given the differences in data between the two sets, we found that label transfer between similar cells was not effective. The approach that we followed was to bridge between datasets. We first computed the nearest neighbour graph (Scanpy neighbours function with k = 30) in each dataset separately, then computed mutual nearest neighbours (MNNs) between the samples using Harmony77. We used the cosine metric to measure the distance between cells in samples, as it is less sensitive to technical artifacts and better reflects biological states in both in and out of the lab. We chose a higher number of mutual neighbours (k = 60) because it is more robust to sparsity in the MNN graph.
Hotspot clusters the gene–gene local correlation matrix into modules using an agglomerative hierarchical clustering procedure that, at each step, merges two genes/modules with the highest pairwise z-scored correlation. Once a merged module contains more genes than a minimum threshold, it is labelled and cannot be combined with other labelled modules. The process ends when the highest pairwise z-score between unmerged modules falls below a minimum value; at that point, all genes that are unlabelled are not assigned to any module.
According to the visualized module trends section, a model is used to fit genes along the Palantir pseudotime. All expression trends for individual genes were calculated on MAGIC-imputed data (see the ‘Gene denoising and imputation’ section), and the s.d. of each expression bin was represented by the s.d. of the residuals of the fit.
To calculate the correlation between the imputed expression and non-intestinal branch probabilities, we compiled known squamous and neuroendocrine cell markers observed in healthy cells or non-CRC cancers. 8a,b We excluded all cells with a probability of <0.5 for a given branch when calculating correlations, to avoid interference by cell states outside that branch. An example of a representative example is shown in the Extended Data Fig., which describes the branches identified in this analysis. 8a). Pearson correlations are used to calculate the non-intestinal branch probabilities in each patient and all of the genes found in the three patients. We found that the top 5 genes ordered by average correlation among the three squamous branches in KG146, KG182 and KG150 are associated with squamous epithelium and keratinization (DMRTA1, NECTIN4, DLX3, CXCL14, LYPD3), and 4 out of the top 5 genes among the three neuroendocrine branches are associated with glial and neural cells (TRPM3, ITPR2, PLPPR1, PPFIA2) (Supplementary Table 5).
For all of the samples in the LARC and TCGA-COAD cohorts, we calculated ssGSEA enrichment scores as described in the ‘ssGSEA analysis’ section using our fetal gene signature as input. We collected samples with ssGSEA enrichment scores greater than 1 s.d. for each cohort and then for lowly enriched samples. below the mean. We performed a log-rank test on DFS between these groups using the Python lifelines (v.0.27.4) package (Extended Data Fig. 7g,h).
We compared our 113-gene fetal signature to previously published dedifferentiation signatures23,34,38,39. For each pair of signatures, we calculated the Jaccard index (number of genes shared between signatures divided by total number of genes in both signatures), demonstrating that existing signatures are clearly distinct from our fetal signature and lack consensus (Extended Data Fig. 7b We also determined how many of the 14 core fetal signature genes (see the next section) are present in each dedifferentiation signature, normalized to the total number of genes in that signature.
The majority of the cells in the first trimester are progenitors and stem cells. By contrast, second-trimester samples consist of mature colon mucosal cell types exclusively, and exhibit strong expression of LGR5, TFF3, SLC26A3, NEUROD1 and POU2F3 (corresponding to ISCs, goblet cells, mature enterocytes, enteroendocrine and tuft cells, respectively). The separation between the first- and second-trimester samples captured the distinction between progenitor-like cell types and colonic crypts.
Source: Progressive plasticity during colorectal cancer metastasis
A Generalised Additive Model with Cubic Splines for an Analysis of Module Score Trends and Enrichment Scores for RNA-Seq Data
We used generalized additive models (GAMs) with cubic splines as smoothing functions as in Palantir (v.1.2)42 to analyse module score trends along DC axes (Fig. 2b and Extended Data Fig. 7a). GAMs improve robustness and reduce sensitivity to density differences, and are useful in capturing non- linear relationships. We fitted trends for a module score using a regression model on the DC values (x axis) and module score values (y axis or colour intensity). The resulting smoothed trend was derived by dividing the data into 500 equally sized bins along the DCs and predicting the module score at each bin using the regression fit. The module score trends from white to the highest saturation were visualized. 7a).
We calculated significance values for these plots using the following strategy: We permuted the labels 1,000 times for each patient, for example, non-Canonical or not non-canonical. For each random permutation we calculated the log-ratios of positive cells with the exception of non-canonical cells. We then performed a rank-sum test comparing our original log-ratios to the combined log-ratios from the random permutations. Our sample distribution may be more than the null, because of the higher incidence of cancer.
For binary clinical features, we separated the enrichment scores into two groups based on the status of the patient from which the bulk RNA-seq data were collected and compared the enrichment scores for each Hotspot module between the two groups using the Mann–Whitney U-test (Fig. 1f,g and Extended Data Fig. 6h–j).
For TCGA, we downloaded and analysed RNA-seq data for 445 tumour samples from the TCGA-COAD study31. RNA raw counts were retrieved using TCGAbiolinks (v.2.26.0)71 and genes with a count of 0 across all the samples were removed, as well as genes that had multiple associated gene symbols or no gene symbol. The VST transformation was performed using the DESeq2 (v.1.38.3) package72. Subsequently, ssGSEA analysis was conducted utilizing the R package GSVA (v.1.46.0)73. In this cohort, 13% of patients have a survival status of alive and an OS follow-up time of <12 months (7% with OS follow-up of <6 months); 0.9% of patients have no new tumour event and a DFS follow-up time of <12 months (0.7% with a DFS follow-up of <6 months).
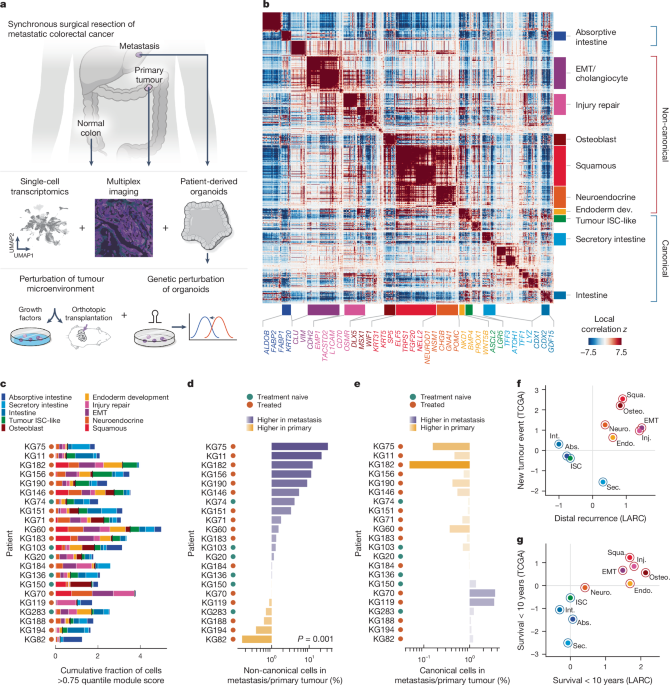
The log- ratios of labelled cell fractions were shown as prevalence in metastatic to patient-matched primary tumours. Canonical and non-canonical module classifications are described in the ‘Hotspot module grouping and annotation’ section. Although cells can show high expression of multiple modules, we found they do not often express their non-canonical modules as highly.
We evaluated the robustness of the Hotspot analysis to the number of HVGs used as input features, based on the consistency of gene autocorrelation, and the consistency of obtained modules. We also evaluated the robustness of the Hotspot modules to bootstrapping of cells.
Source: Progressive plasticity during colorectal cancer metastasis
Differential expression analysis of epithelial differentiation, mucus production and small-molecule transport in the intestinal gastrointestinal tract using Scanpy and MAST
Canonical: modules describing canonical intestinal cell types and processes such as epithelial differentiation, mucus production and small-molecule transport, which are critical for maintaining normal intestinal function.
We focused on 23 of the modules that represented meaningful biological genes and did not examine 14 modules annotated as cell cycle/ proliferative (2 modules), cell stress (4 modules), and cilia (1 module) or anything else. We took the same biological interpretation for all modules within a group, and then manually grouped 19 modules into 6 groups, to make sure the local correlations between genes of grouped modules were high. 5a,b). The final genes resulted from six grouped modules and four single modules. 5a,b and Supplementary Table 4).
We performed differential expression analysis of ISC cells against all untreated tumour cells using MAST (v.1.16.0) and GSEA using relevant cell type gene sets from the literature (Supplementary Table 3) as well as all Hallmark66 and KEGG67 gene sets (Extended Data Fig. 3f). The Python package gseapy had 10,000 permutations and the default parameters in its prerank function, which was used for GSEA.
The cells we identified were in the epithelial compartment. Evidence of copy-number alterations is compared with cells from non-tumour colon samples and clustering is distinct from non-tumour cells.
We partitioned the clusters into immune compartments based on marker gene expression. 3a,b). The score_genes function in Scanpy was used to score expression of the genes from the ref. 61, similar to the strategy used in that study (signatures for each compartment are shown in Supplementary Table 7). The compartment had a maximal score and the cluster was assigned to it.
The Scanpy was used to generate the Gene signature scores in our study. score_genes function, which calculates the mean expression of genes of interest subtracted by the mean expression of a random, expression-matched set of reference genes. To account for expression-level differences across genes within signatures, we provided z-normalized expression data as the input for this function.
OCT-embedded tissue and FFPE tissue samples were sectioned and placed on a Visium Spatial Gene Expression Slide following Visium Spatial Protocols–Tissue Preparation guide. Following the user guide, Visium Spatial Gene Expression Reagent kits were constructed using tissue samples. Clinical MSK-IMPACT targeted exon sequencing was performed on tumour/normal tissue from 27 out of 31 patients and revealed expected mutations.