A large amount of people from Mexico City have genotyping and genotyping done
Inferring Indigenous ancestry from the Americas and the MXB Project using MAAS-MDS64: a clustered approach using GNOMIX3
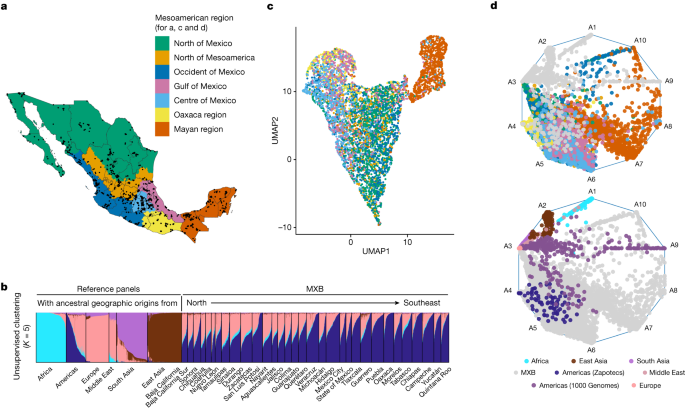
For the MAAS-MDS64 analyses, we used GNOMIX3 for local ancestry inference using its preset ‘best’ mode and then masked the non-Indigenous segments. For the European reference, we used the cohorts Iberian populations in Spain (IBS) and British from England and Scotland (GBR) from 1KGP (198 samples)40, for ancestries from Africa, the Yoruba in Ibadan, Nigeria (YRI) cohort from 1KGP (108 samples), and for ancestries from the Americas, Peruvian in Lima, Peru (PEL) from 1KGP (only those samples with >95% ancestry from the Americas) and the 50 genomes of Indigenous individuals across Mexico generated as part of the MXB Project (79 samples)65. For the PEL, we relied on an analysis with a clustering technique that gave us samples with an assignment to a different cluster than what they were shared with. The 50 new genomes from MXB were selected to have high Indigenous ancestries. The reference genomes were merged with each array resulting in 856,352 SNPs in array 1 and 967,338 in array 2. Array 1 included 10 Indigenous groups from NMDP genotyped with the Affymetrix 6.0 array: Tarahumara, Huichol, Purepecha, Nahua, Totonac, Mazatec, Northern Zapotec (from Villa Alta district, Northern Sierra in Oaxaca state), Triqui, Tzotzil and Maya (from Quintana Roo state). Array 2 included the 6,051 individuals from the MXB project genotyped with MEGA. The MAAS-MDS was applied to the Indigenous American ancestry segments to hide intercontinental parts of African and European origin. The analysis was run using average pairwise genetic distances and considering only individuals with >20% Indigenous American ancestries, to generate ancestry-specific MDS axes for ancestries from the Americas in the MXB (Supplementary Fig. 24).
Due to the demographic and environment histories that we are interested in, such regions are only useful for our analyses. This is only one arbitrary scale to discretize at, and we also consider the origins and implications of ancestral variations within such regional groupings in several analyses, in which we carry out dimensionality reduction within such regional groupings (for example, MDS1(A(Americas)) and MDS2(A(Americas))).
The ABCA1 variant frequencies were computed using plink in a sample of individuals who were from the MXB.
Physical positions of imputed variants were mapped from genome build GRCh37 to GRCh38 using the program LiftOver, and only variant positions included on the Affymetrix GSA v.2 were retained. After further filtering out variants with imputation information r2 < 0.9, the following QC steps were performed before merging of the MAIS Affymetrix and Illumina datasets: (1) removal of ambiguous variants (that is, A/T and C/G polymorphisms); (2) removal of duplicate variants; (3) identifying and correcting allele flips; and (4) removal of variants with position mismatches. Merging was performed using the –bmerge command in Plink (v.1.9).
The relationship assignments and relatedness-inference criteria were based on the kinship coefficients and IBD share from the KING software. To reconstruct all of the first degree family networks, we took the estimates of relatedness, sex and age of each individual into account. The first-degree family networks were reconstructed in 99.3% of the way. Graphviz was used to create networks such as the ones presented in Supplementary fig. 5. We used the sfdp layout engine which uses a ‘spring’ model that relies on a force-directed approach to minimize edge length.
Fixed predictors of complex trait variation are genetic and environmental variables. Polygenic scores were computed with UKB summary statistics, genetic ancestries were estimated with MAAS-MDS, and the amount of ROH was included. We also considered biogeographical variables such as latitude, longitude and altitude (metres). We considered the demographic variables of their age and sex. Last, we considered sociocultural variables: educational attainment (which shows a positive correlation with income levels (Supplementary Fig. 52); however, income levels are available only for a third of the individuals); whether they speak an Indigenous language or not as a proxy for differential experience of discrimination and culture; and whether they live in an urban or rural environment. BMI was included as a covariate for all quantitative traits except height, BMI and creatinine (Fig. 5 and Supplementary Figs. 53–58). To ease interpretation of the mixed-model coefficients for jointly considered numeric and binary predictors, we standardized predictor variables as follows74. To make coefficients of the predictors comparable to those for untransformed predictors, we divide each variable by its standard deviation. The predictors were narrowed down to thebinary andnumeric. The significance of the covariates is demonstrated by their use in the full model.
In order to measure quantitative aspects, they were measured. Data was removed from outliers with apparent errors in data entry and negative values based on distribution density. Participants with a measurement between 100 and 200 cm were limited in their height and weight. The values that were greatly different from those taken at the time of the survey were removed. The measurement was based on the number of people who had eaten in the 8–12 h before the samples were taken.
Biometric data were filtered to remove outliers with apparent errors in data entry. Outliers were identified on the basis of distribution density over the complete dataset of >6,000 individuals, resulting in height between 100 and 200 cm and weight between 25 and 300 kg.
An anthropological and archaeological context in the Mesoamerican regions of Mexico: AMR data visualization and data imputation for topmed 10k and MCPS 10k
Blood pressure was manually curated for individuals for whom values differed by more than 20 units for the two readings taken, for whom diastolic pressure was higher than systolic, or for whom values were unusually high or low (<30 or >300). Both readings were checked manually and the readings that differed were discarded. The remaining samples were merged with the updated values. A set of adjusted blood pressure phenotypes was also generated, adjusting for treatment for hypertension. 15 units were added to systolic blood pressure and 10 units to diastolic blood pressure for those who were reported to be receiving hypertension treatment.
Every individual has access to data with regard to various factors such as access to healthcare and clean water, yearly income, educational attainment and even if they don’t speak an Indigenous language.
Localities were assigned values of latitude, longitude and altitude (metres) using data from the National Institute of Statistics and Geography (INEGI) in Mexico.
We used an anthropological and archaeological context to delineate different Mesoamerican regions10. An individual’s locality was used to place them into one of the seven regions: the north of Mexico, the north of Mesoamerica, the centre, occident and Gulf of Mexico, Oaxaca and the Mayan region in the southeast10. This classification was used to visualize and regionalize some of the population structure and history analyses, especially those relating to Indigenous genetic substructure within Mexico.
The 1000 Genomes genes were downloaded into the database and then sorted by coverage. We used only those 490 AMR samples in the MXL, CLM, PUR and PEL subpopulations. The Illumina HumanOmni ExpressExome and Illumina GSA were used to build the two subsets of genotypes that we used for the TOPmed and MCPS10k imputation programs.
Uniform manifolds approximation and projection analysis using smartpca and the Admixture Bayesian method. I. UMAP and FST analysis
It was smartpca that carried out the PCA. The uniform manifolds approximation and projection (UMAP) analysis was carried out with principal components generated by smartpca. FST analysis was carried out using smartpca.
Mutation burden is defined as the sum of derived alleles carried by an individual. A computational pipeline using vcftools, python, linux and R was used to compute mutation burden in different classes of variants, and at different derived allele frequency thresholds. Considering all the allele frequencies we computed a rare mutation burden or an overall one. The R packages were used in ourpipeline. The global ancestry percentage from different current-day continental origins in all individuals were correlated with the mutations burden. The ancestry estimates were from the admixture analysis. We computed Spearman’s correlation and P value.
The events that took place in the MXB have been inferred using AdmixtureBayes. We used the default parameters, which included the number of chains and iteration, to ensure convergence and also set the flag to the lower value. We plotted the tree with the highest posterior probabilities, which provides a visual representation of the inferred admixture events and allows us to explore the uncertainty in the inferences. There are further details of the AdmixtureBayes method in the paper.
Creating Complex Trait Variation: A Mixed-Model Analysis of Lead Associations and Potential Causal Groups for lme4qtl
To assess the factors involved in creating complex trait variation, we carried out a mixed-model analysis using the lme4qtl R package for all quantitative traits. lme4qtl allows flexible model creation with multiple random effects73.
Variants were annotated as previously described38. The Ensembl variant effect predictor can be used to predict the consequences of each variant chosen across all transcripts. We derived transcript annotations from a combination of MANE, APPRIS and Ensembl tags. APPRIS had the highest priority followed by MANE. The code of the Ensembl transcript was used when there were no MANE or APPRIS annotations for the gene. Gene regions were defined using Ensembl release 100. Predicted loss-of-function variants are those that are annotated as stopping, start lost or frameshift and which the alleles of interest were not the ancestral ones. Five annotation resources were utilized to assign deleteriousness to missense variants: SIFT; PolyPhen2 HDIV and PolyPhen2 HVAR; LRT; and MutationTaster. Missense versions were considered ‘probably deleterious’ if predicted in a way that was likely benign or possibly deleterious if predicted in a way that was likely deleterious.
Lead association SNPs and potential causal groups were defined using FINEMAP (v1.3.1; R2 = 0.7; Bayes factor ≥ 2) of SNPs within each of these regions on the basis of summary statistics for each of the associated traits70. FUMA SNP2GENE was then used to identify the nearest genes to each locus on the basis of the linkage disequilibrium calculated using the 1000 Genomes EUR populations, and explore previously reported associations in the GWAS catalogue40,71 (Supplementary Table 7).
Development and Storage of a Novel Blood Sample for Studying the Health Effects of Smoking in Mexico Using UV-VIS Spectroscopy and DNA Library Amplification
In the late 1990s, scientists at the UNAM and University of Oxford began discussing how to measure the health effects of tobacco in Mexico. There is a plan to establish a prospective cohort study that would investigate the effects of tobacco on other factors, including factors measurable in the blood. There were more than 100,000 women and 50,000 men that agreed to take part in the study, which included physical measurement, a blood sample and the possibility of cause-specific mortality. More women were recruited because the visits were mainly made during working hours when women are most likely to be at home, and more men were recruited because visits were extended into the early evenings and weekends.
A transport box of ice packs was used to chill the blood sample for transfer to the central laboratory. The samples were refrigerated overnight at -4 C and then centrifuged the following day at 4 C. Plasma and buffy-coat samples were stored locally at −80 °C, then transported on dry ice to Oxford (United Kingdom) for long-term storage over liquid nitrogen. DNA was extracted from buffy coat at the UK Biocentre using Perkin Elmer Chemagic 360 systems and suspended in TE buffer. UV-VIS spectroscopy using Trinean DropSense96 was used to determine yield and quality, and samples were normalized to provide 2 μg DNA at 20 ng μl–1 concentration (2% of samples provided a minimum 1.5 µg DNA at 10 ng µl–1 concentration) with a 260:280 nm ratio of >1.8 and a 260:230 nm ratio of 2.0–2.2.
Genomic DNA samples were transferred to the Regeneron Genetics Center from the UK Biocentre and stored in an automated sample biobank at –80 °C before sample preparation. DNA libraries were created by enzymatically shearing DNA to a mean fragment size of 200 bp, and a common Y-shaped adapter was ligated to all DNA libraries. Unique, asymmetric 10 bp barcodes were added to the DNA fragment during library amplification to facilitate multiplexed exome capture and sequencing. All samples were captured on the same lot of oligo-nucleotides after pooling equal amounts of sample. Quantitative PCR is used to amplify and quantify the captured DNA. The multiplexed samples were pooled and then sequenced using 75 bp paired-end reads with two 10 bp index reads on an Illumina NovaSeq 6000 platform on S4 flow cells. A total of 146,068 samples were made available for processing. The majority of the 2,628 samples didn’t go through due to low or no DNA being present. A total of 143,440 samples were sequenced. The average 20× coverage was 96.5%, and 98.7% of the samples were above 90%.
The MCPS WES and WGS data were reference-aligned using the OQFE protocol35, which uses BWA MEM to map all reads to the GRCh38 reference in an alt-aware manner, marks read duplicates and adds additional per-read tags. The OQFE protocol retains all reads and original quality scores such that the original FASTQ is completely recoverable from the resulting CRAM file. The DeepVariant produced a gVccF for each input OQFE CRAM file, using the default WGS and custom exome parameters. The gVCFs were aggregated using a method called GLnexus. The steps of this protocol were executed using open-source software.
We implemented a supervised machine- learning algorithm to discriminate between high and low quality variants. We defined a set of positive control and negative control variations on the basis of a number of criteria, including concordance in calls between array and exome-sequencing data, transmitted singletons, and likely high quality sites. To define the high-quality set, we first generated the intersection of both of theVariants that passed in the gnomAD v. 3.1 genomes. The 1000 Genomes project and a previous study both have gold-standard insertions and deletions available through the GATK resource bundle. To define the external low-quality set, we intersected gnomAD v3.1 fail variants with TOPMed Freeze 8 Mendelian or duplicate discordant variants. The control set of variants was binned by allelic frequencies and then randomly sample such that they retained an equal number of variant in each bin. A support machine used a radial basis function was trained on up to 33 available site quality metrics which included median values for allele balance in Heterozygote calls and whether a variant was split from a multi-allelic site. We split the data into training (80%) and test (20%) sets. We used a grid search with fivefold cross- validation to find hyperparameters that had returned the highest accuracy, then applied it to the test set to confirm accuracy. This approach identified a total of 616,027 WES and 22,784,296 WGS variants as low-quality (of which 161,707 and 104,452 were coding variants, respectively). We further applied a set of hard filters to exclude monomorphs, unresolved duplicates, variants with >10% missingness, ≥3 mendel errors (WGS only) or failed Hardy–Weinberg equilibrium (HWE) with excess heterozgosity (HWE P < 1 × 10–30 and observed heterozygote count of >1.5× expected heterozygote count), which resulted in a dataset of 9,325,897 WES and 131,851,586 WGS variants (of which 4,037,949 and 1,460,499 were coding variants, respectively).
We used Shapeit (v.4.1.3; https://odelaneau.github.io/shapeit4) to phase the array dataset of 138,511 samples and 539,315 autosomal variants that passed the array QC procedure. To improve the phasing quality, we leveraged the inferred family information by building a partial haplotype scaffold on unphased genotypes at 1,266 trios from 3,475 inferred nuclear families identified (randomly selecting one offspring per family when there was more than one). We then ran Shapeit one chromosome at a time, passing the scaffold information with the –scaffold option.
The allele frequencies were estimated in two steps. We built a graph based on the IBD sharing at a locus. We estimated allele counts and allele numbers by looking at the component parts of the IODB graph. Our approach is very similar to that of DASH. However, we make different assumptions about how errors affect the IBD graph and additionally compute ancestry-specific frequencies using local ancestry inference estimates.
IBD segments from hapIBD were summed across pairs of individuals to create a network of IBD sharing represented by the weight matrix (W\in {{\mathbb{R}}}{\ge 0}^{n\times n}) for n samples. Each entry ({w}{{ij}}\in W) gives the total length in cM of the genome that individuals i and j share identical by descent. We wanted to create a low-dimensional visualization that looked like the IBD network. We used a similar approach to that described in ref. 14, which used the eigenvectors of the normalized graph Laplacian as coordinates for a low-dimensional embedding of the IBD network. The degree matrix for the graph is (d_ii=sum _jw_ij) and 0 elsewhere. The identity matrix in the Laplacian graphs is defined to be (D-1W), where I am.
We imputed the filtered array dataset using both the MCPS10k reference panel and the TOPMed imputation server. For TOPMed imputation, we used Plink2 to convert this dataset from Plink1.9 format genotypes to unphased VCF genotypes. For compatibility with TOPMed imputation server restrictions, we split the samples in this dataset into six randomly assigned subsets of about 23,471 samples, and into chromosome-specific bgzipped VCF files. Using the NIH Biocatalyst API (https://imputation.biodatacatalyst.nhlbi.nih.gov), we submitted these six jobs to the TOPMed imputation server. We used bcftools to join the VCFs after the jobs were done. We used Impute5 for the MCPS10k imputation. The imp5Chunker program requires a minimum window size of 5 kbyte and a minimum buffer size of 500 kb. Information scores were calculated using qctool (https://www.well.ox.ac.uk/~gav/qctool_v2/).
mathopsum limits_i=1nmathopsum
Source: Genotyping, sequencing and analysis of 140,000 adults from Mexico City
Estimation of effective sample size for exome singleton sites in the UK based on an ancestry comparison in MCPS and TOP Med
An estimate of the effective sample size for population k at the site is ({n}{k}={{\rm{AN}}}{k}/2). Singleton sites can be hard to phase using existing methods. Family information and phase information in sequencing reads was used in the WGS phasing, and this helped to phase a proportion of the singleton sites. We found that 42% of exome singleton were located in stretches of their parent’s ancestry. For these variants, we gave equal weight to the two ancestries when estimating allele frequencies.
Jle 2,rand,1le. Ile N E_l,(I,j)_l
$$\begin{array}{l}W={\sum }{C\in {C}{{\rm{ALT}}}}{\bar{p}}{C}\ N={\sum }{C\in {C}{{\rm{ALT}}}}{\bar{p}}{C}+{\sum }{C\in {C}{{\rm{REF}}}}{\bar{p}}_{C}\end{array}$$
To avoid overfitting with respect to selection of a PRS algorithm and its associated tuning parameters, LDpred (https://github.com/bvilhjal/ldpred) with ρ value of 1 was chosen from a recent publication of BMI and obesity27. Summary statistics were restricted to HapMap3 variants and followed existing filtering recommendations. The imputed variant was obtained from either the MCPS10k reference panel or the TOP Med panel. The UK biobank data has different values for Africa, East Asian, European, Latino, and South Asian.
As part of the MXB Project, the 50 genomes of indigenous individuals across Mexico were generated as part of the MXB Project. The analysis was run using average pairwise genetic distances and considering only individuals with >20% Indigenous American ancestries. The analysis was run using average pairwise genetic distances and considering only individuals with >20% Indigenous American ancestries.